·综 述·
【摘要】 肌萎缩侧索硬化(amyotrophic lateral sclerosis, ALS)是一组以选择性上、下运动神经元损害为特征的致死性神经变性疾病,患者多在3~5年发展为呼吸衰竭死亡。本文对国内外关于ALS相关基因及临床特点、基因型-表型用于临床诊断、基因研究新进展以及基因治疗进行综述。
【关键词】 肌萎缩侧索硬化; 基因型; 临床特点; 基因诊断; 基因治疗
肌萎缩侧索硬化(amyotrophic lateral sclerosis, ALS)是缓慢进展性运动神经元疾病,主要累及大脑皮质、脑干、脊髓前角等运动神经元,最终发展为呼吸衰竭,80%~ 90%的患者于发病后3~5年死亡。该病迄今缺乏有效治疗,目前唯一被美国FDA批准的临床药物利鲁唑(riluzole)也仅能延长患者的生存期平均3个月以上[1]。根据其发病和遗传特点,可分为散发性肌萎缩侧索硬化(sporadic amyotrophic lateral sclerosis, SALS)和家族性肌萎缩侧索硬化(familial amyotrophic lateral sclerosis, FALS),其中FALS约占全部病例的5%~10%。现就国内外关于ALS相关基因和临床特点的研究及进展综述如下。
目前至少发现了24种与FALS相关的基因突变,其中部分也发现存在于SALS中。这些基因突变发生率中较高的主要是: C9ORF72、SOD1、FUS和TARDBP(trans-activation response DNA-binding protein 43, TDP43),占全部FALS的60%[1],而占SALS的10%~11%。已发现的FALS的临床亚型有: ALS1-13、ALS合并包涵体肌炎、ALS15-21、ALS合并额颞叶痴呆FTD1(front temporal dementia, FTD)及ALS合并额颞叶痴呆FTD2等两种亚型。
ALS1由SOD1基因突变所致。ALS2由编码GTP酶调节子的Alsin基因突变引起核内体动力学改变所致,是一种少见的常染色体隐性遗传的青少年型ALS,以选择性的上运动神经元缓慢进行性的退变、伴或不伴下运动神经元损害为特征,神经肌肉损害严重。较少报道的ALS3基因成人起病,病变部位和临床表现均与ALS2相同,但病程可长达10年[2]。编码DNA/RNA解旋酶的SETX基因突变所致ALS4为青少年型FALS,呈常染色体显性遗传,起病年龄常小于25岁,以肢体远端肌无力、肌萎缩和锥体束征阳性为主要临床特征,进展缓慢,患者可有正常寿命。SPG11基因突变可同时出现在ALS2和ALS5上,ALS5以下运动神经元缓慢进展的肌无力和肌萎缩为特征,常沿四肢末端到延髓进展。ALS6的致病基因为FUS基因,ALS7常染色体显性遗传的成人型FALS有关的突变基因仍有待确证,临床表型尚无统一说法[3]。小囊泡相关膜蛋白(vesicle-associated membrane protein-associated proteinB, VAPB)基因突变的ALS8临床表现为典型的ALS或迟发性脊肌萎缩症,成人起病,呈常染色体显性遗传,特点是下运动神经元受累为主,常伴有肌束震颤、姿势性震颤和痛性痉挛。血管生长素(angiogenin, ANG)基因所致ALS9患者成人起病,呈常染色体显性遗传,临床表现与典型的ALS相同,外显率低[4]。Tau基因所致ALS10,FIG4基因突变是常染色体显性遗传的FALS即ALS11和SALS的共同致病基因,表现为遗传性痉挛下身瘫痪、原发性侧索硬化、人格改变。视神经病变诱导反应蛋白(optineurin, OPTN)基因所致ALS12既可表现为常染色体显性遗传又可表现为常染色体隐性遗传。ALS13由共济失调蛋白-2(ataxin 2, ATXN2)基因其突变所致,呈常染色体显性遗传性脊髓小脑性共济失调,是一组异质性的神经退行性疾病,其特点是渐进的小脑、脑干和脊髓的退行性变,结合视神经萎缩、眼肌麻痹、延髓和锥体外系体征、周围神经病变和老年痴呆症等临床特点。ALS14,即ALS合并包涵体肌炎,由含缬酪肽蛋白(valosincontaining protein, VCP)基因突变所致,较少见表现为原发性侧索硬化、进展性球麻痹、恶性进展、舞蹈手足徐动症样动作、痉挛性瘫痪等[3]。泛素-2(ubiquilin 2, UBQLN2)基因突变破坏了蛋白质的降解过程,导致一种罕见的X连锁显性遗传的ALS15发病[5]。SIGMAR1基因其所致的ALS16呈常染色体隐性遗传,患者1~2年内即可出现运动神经元疾病,临床表现为呈缓慢进展的肌萎缩、肌无力等,没有认知障碍[6]。ALS17有CHMP 2B突变所致,上运动神经元为主进展性肌萎缩、帕金森样症状伴额颞叶痴呆。肌动蛋白-1 (pentacyclictriterpene synthase 1, PFNl)基因其所致的ALS18呈常染色体显性遗传[7]。ALS19由ERBB4基因突变所致,累及延髓,呼吸道。ALS20中发现hnRNPA1基因突变,表现为佩吉特病、包涵体肌病。ALS21则可见MATR3基因突变,累及延髓,可见末端肌病(包涵体肌病)。ALS-FTD1可由C9ORF72基因编码区障碍导致。ALS-FTD2则见CHCHD10和TBK1基因突变[3]。ALS20和ALS-FTD亚型均可见伴有明显的额颞叶痴呆。
1.1 ALS1: 超氧化物歧化酶1(SOD1)基因
位于21号染色体长臂上的SOD1基因,其通过编码的蛋白质超氧化物歧化酶(SOD)参与致病。目前约12%的FALS和 1%~ 2%的SALS与致病基因铜锌SOD1突变有关,SOD1基因突变导致蛋白毒性功能获得而不是功能丧失有关。临床特点为: (1) 突变呈一定的地域分布特点,如A4V突变所致的以下运动神经元受累为主特点,生存期较短,在北美最常见;D90A突变在欧洲最常见;H46R突变所致的以一侧下肢无力起病,逐渐累及对侧下肢,进展十分缓慢,生存期长,预后良好,仅见于亚裔[8]。(2) 大部分的SOD1基因突变呈常染色体显性遗传,但D90A突变既呈常染色体显性遗传,也呈常染色体隐性遗传。(3) 各突变临床表型有差异: D90A同型突变表现为下肢慢性进展性瘫痪,随后累及上肢、胸腔和延髓肌群,伴有非典型的非运动损伤,如共济失调、神经痛,酸痛感、热感和膀胱障碍;G72C突变以对称性近端无力及腹部无力起病,起初肌电图正常,较快进展到上肢和延髓,较早发病,病程进展迅速,生存时间只有1年;E133V突变以肌束震颤和四肢肌肉痉挛起病,体检有上下运动神经元受损的表现,后累及延髓,中年发病,病程进展快速,生存时间为4年[9]。
1.2 ALS6: 融合肉瘤/异位脂肪肉瘤(FUS/TLS)基因
FUS基因编码的突变导致蛋白翻译错误可能成为ALS6的致病机制。临床特点为: (1) ALS6残留的神经元和增生的神经胶质细胞胞质中可见特征性的FUS蛋白免疫反应阳性的嗜酸性包涵体,并呈现弥散的泛素阳性的免疫染色,周围巨噬细胞浸润。(2) 呈常染色体显性遗传,平均在45岁起病,病程进展较快,平均存活期为5年。P525L突变发病年龄较其他突变早(13~25岁),病情进展更快,病程为6~20个月[10]。(3) 临床表现呈现经典的ALS,从右下肢起病,逐渐进展为四肢远端肌肉无力萎缩,腱反射活跃,肌电图为失神经支配,而很少伴有其他非典型症状,如延髓肌症状、认知改变等。(4) 不同突变类型的FUS突变的临床表型存在较大的差异,最常见的R51C突变主要表现为对称性的肢体近端肌肉无力,多累及躯干肌,后期才累及肢体远端,较少出现上运动神经元受累的表现,可累及颈屈肌、伸肌无力导致垂头综合征[11]。
1.3 ALS10: TAR DNA结合蛋白(TDP43)基因
TARDBP基因突变导致神经细胞中TDP43蛋白正常功能的缺失和异常功能的获得。其基因突变与大约4%的FALS病例和1%散发性病例有关,总的突变率为5%。所致的ALS10的临床表型为常染色体显性遗传的经典成人型ALS,起病早,病程长,中位生存期大约为3年,以单侧肢体受累起病,主要表现为上肢起病(60.7%),渐向四肢发展,可伴有帕金森样症状、舞蹈样动作、进展性核上性球麻痹,而少见额颞叶痴呆[3]。
1.4 ALS-FTD1: 9号染色体开放阅读框(C9ORF72)基因
C9ORF72基因突变是欧洲人群发生ALS最常见的突变基因,也是高加索人群中最常见的,其他人群中则比较少见[2]。其突变所致的ALS-FTD为罕见的FALS,发病率<5%,是常染色体显性遗传,临床特点为: (1) 好发于45~65岁年龄,病程长,年轻起病的ALS合并FTD的风险增加;(2) 延髓起病、进展迅速,无力症状最先易累及球部肌肉,上肢无力症状亦重于下肢,部分患者在疾病晚期仍可保留行走能力;(3) 患者本人或者其近亲很可能同时伴有认知改变甚至痴呆;(4) 可表现为帕金森样综合征,还可表现为橄榄脑桥小脑变性、非典型帕金森样综合征和皮质基底节综合征等,或者合并多发性硬化;(5) 临床表型在家族患者内部也可具有临床异质性;(6) 预后较单纯FTLD和单纯ALS更差,其中位数生存期较二者分别缩短4年和1年左右;(7) 影像学表现以双侧额颞叶局限性脑萎缩为典型改变,代谢影像学呈双侧额叶为主要低代谢区;(8) 假性球麻痹及球部起病均是ALS合并认知功能障碍的危险因素[10]。
目前认为SALS属于复杂因素疾病,其易感基因包括: NEFH、APEX、ANG、HFE、PON、SOD、PRPH、CYP、EAAT2、LIF、SMN2和VEGF等。这些基因中,NEFH、APEX和ANG被较多报道[12]。
现今并无诊断ALS致病基因的标准程序,但结合考虑青少年起病、临床特点、疾病进展和遗传特征等因素,可以找到可能的致病基因。本文结合ALS的基因型与临床表型之间的关系,总结出了ALS诊断流程图(图1~2),可为临床确诊提供新思路[9,12]。
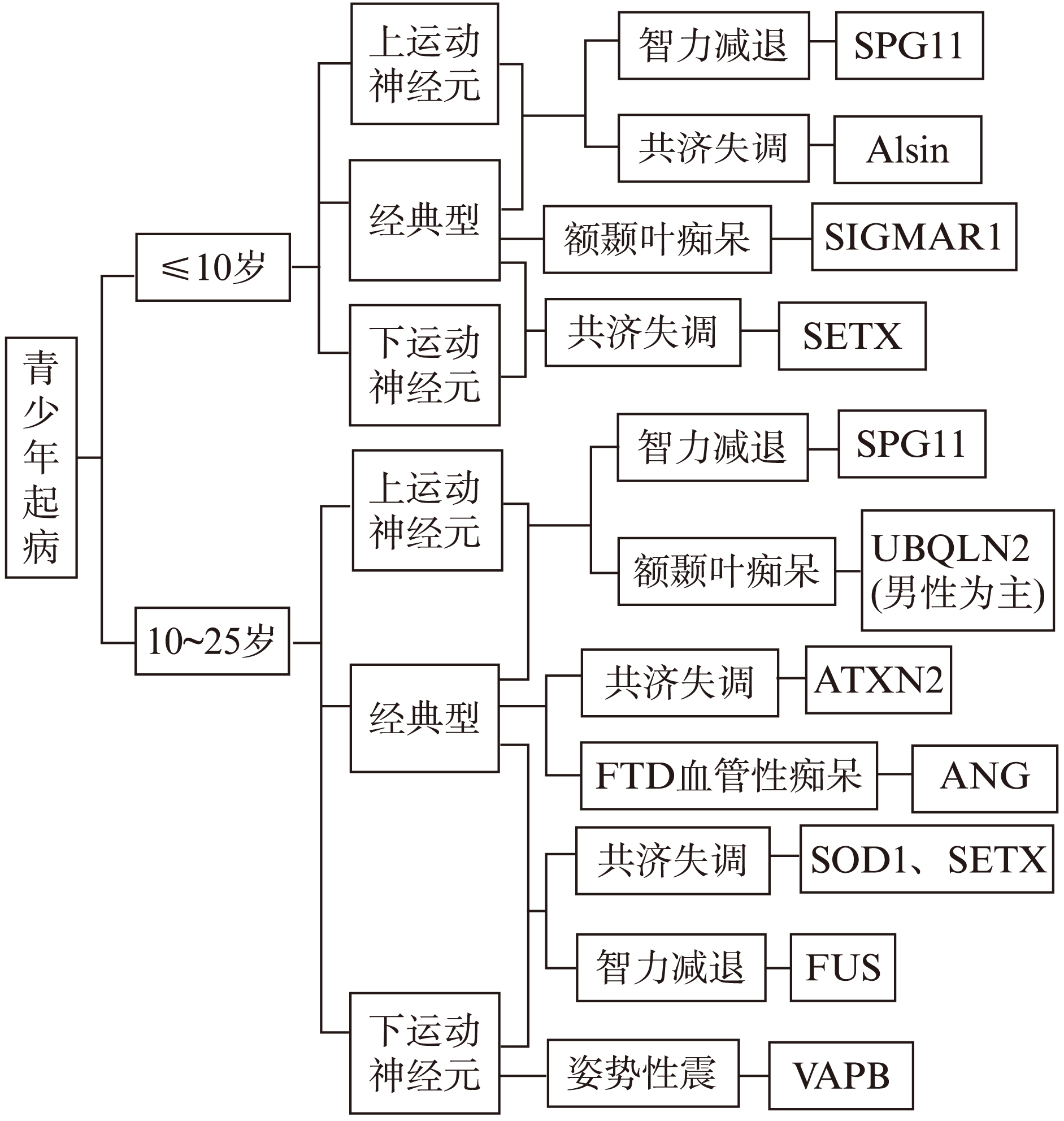
图1 以青少年起病为特征的ALS诊断流程图
Fig.1 Flow chart for diagnosis of hereditary juvenile-onset ALS
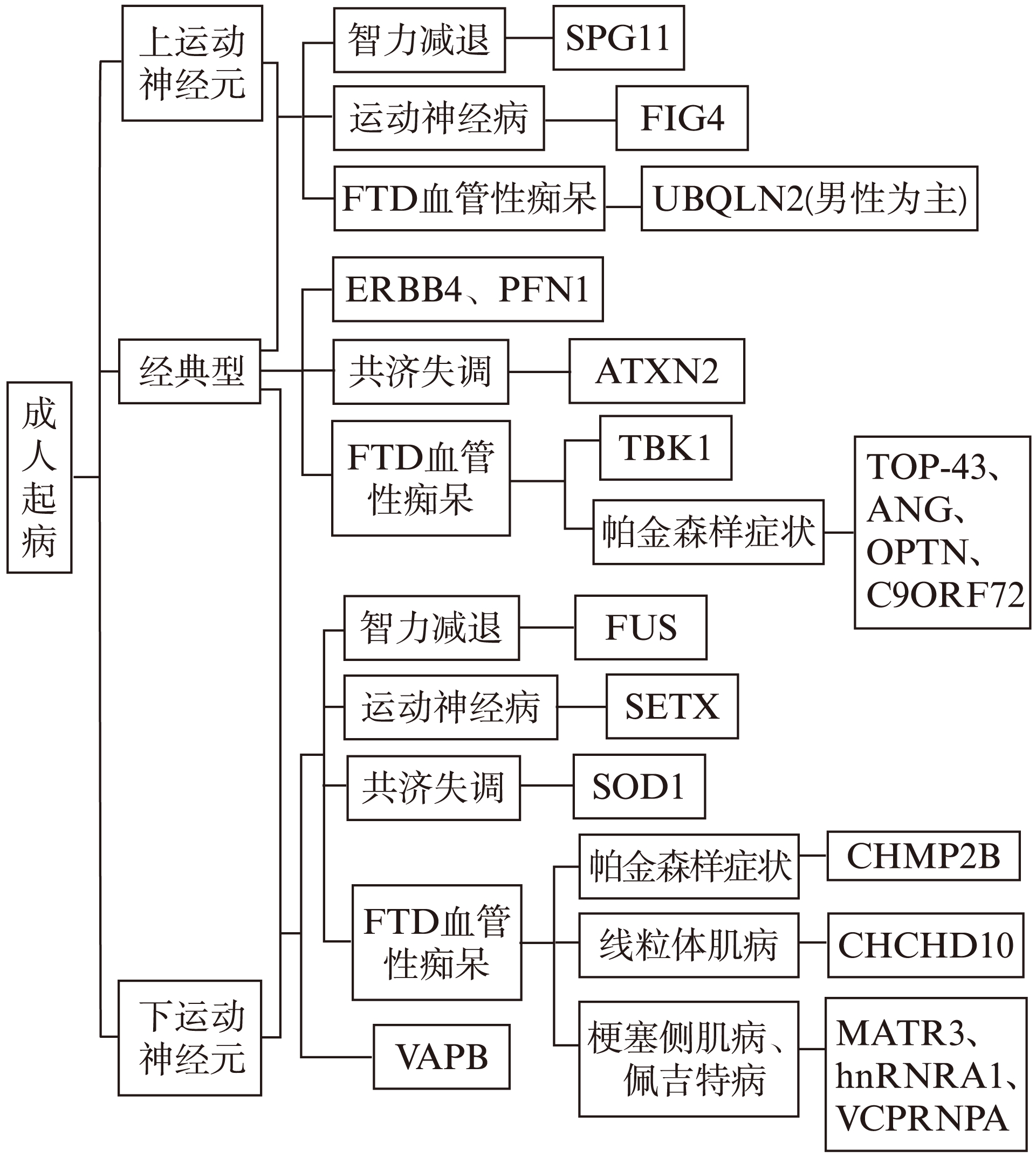
图2 以成年起病为特征的ALS诊断流程图
Fig.2 Flowchart for diagnosis of hereditary adult-onset ALS
4.1 锌指蛋白与其下游转化生长因子-β
在ALS患者中检测到锌指蛋白512B(zinc finger protein 512B, ZNF512B)基因表达水平以及血液中TGF-β表达减少,随之引起神经元保护作用减弱。因此推测表达ZNF512B基因的个体,患病风险增高、病程进展迅速、临床预后较差[13]。
4.2 自噬相关基因
细胞自噬减少引起异常蛋白聚集到神经元,如β淀粉样蛋白、Hunting蛋白等,进而导致运动功能及记忆认知功能等的损害。研究报道[14]在ALS转基因鼠的海马组织中,自噬相关基因微管相关蛋白轻链3-Ⅱ(Microtubule-associated protein 1A/1B-light chain 3Ⅱ, LC3-Ⅱ)的蛋白表达水平明显上调,表明其诱导的自噬激活可能参与了ALS发病。同时,转录因子EB(transcription factor EB, TFEB)及下游自噬相关基因Beclinl在ALS转基因鼠海马中也表达异常。TFEB是调控溶酶体生物合成的主要基因,进而参与调节细胞自噬。在ALS发病早期,TFEB及Beclinl表达上调,表明自噬增强,自噬体和溶酶体活动增强,从而增强其清除神经元中异常蛋白的作用;在ALS发病中晚期,TFEB和Beclinl表达下调,表明自噬减弱,从而不能清除神经元中多余的蛋白质,导致SOD l蛋白等在神经元异常聚集,引起神经元变性、死亡。
近年来随着ALS基因研究不断进展,该病许多致病基因已被鉴定出来,基因治疗为治疗ALS提供新的策略。
从致病机制角度来说,ALS治疗面临的一个主要问题是其复杂的致病机制,包括运动神经元选择性死亡、兴奋性氨基酸毒性、线粒体和内质网功能障碍、RNA合成障碍、轴突运输障碍、蛋白质合成和聚集异常、氧化应激反应、细胞凋亡、神经纤维异常和炎症因素等。抗凋亡蛋白的研究发现,SOD1G93A转基因鼠体内外转染Bcl-xl和Bcl-2基因能对运动神经元细胞起到保护作用[15]。抗氧化应激反应的研究发现,在ALS细胞模型中转染过氧化还原酶3(peroxiredoxin 3, PRDX 3)和核因子2相关因子2(NF-E2-related factor 2, NRF 2)等抗氧化基因能发挥一定治疗作用[16]。过表达抗SOD1蛋白基因的腺病毒6(AAV-6)转染初生SOD1G93A转基因小鼠全身肌肉,可阻止肌肉萎缩但不能阻断疾病的进展,或许与中枢神经系统AAV-6低转染率和血脑屏障低穿透率有关。过表达D3H5抗体单链的AAV转染SOD1G93A模型鼠后,能够减少错误折叠的SOD1蛋白毒性,脊髓中异常折叠的SOD1蛋白减少,随之的神经元异常反应也减少[17]。抑制小胶质细胞中突变基因的表达时,可显著降低发病后疾病进展速度。另外,SOD1G93A鼠脊髓内转染过表达IL-10 AAV证实有效[18]。
从基因突变角度来说,(1) SOD1基因: 内源性SOD1敲除治疗ALS研究已在部分研究中证明是安全有效的。在体外培养的细胞中,RNAi介导的SOD1基因沉默可延长SOD1小鼠的寿命[16]。将作用于SOD1基因的稳定的siRNA注入脑脊液中能敲除50%的靶mRNA从而发挥作用[19]。一种以反义寡核苷酸为基础可抑制SOD1产生的药物也正在进行第一阶段的研究以评估安全性(NCT01041222)[20]。(2) TDP43基因: 次黄嘌呤腺苷脱氢酶(adenosinedeaminasesthatacton RNA, ADAR 2)调节Ca2+内流增加从而导致神经元死亡,ADAR2 mRNA是TDP43蛋白的靶RNA,TDP43蛋白在ADAR2的表达中起调节作用,TDP43基因异常常伴随ADAR2在中枢神经系统表达异常。Yamashita等[21]在ADAR2敲除鼠中转染过表达ADAR2的AAV后发现运动神经元TDP43表达正常,运动损伤进展被阻止。同时,研究发现在TDP43模型鼠中出现的上肢前臂瘫痪能在转染人类无义介导mRNA降解途径(nonsense-mediated mRNA decay, NMD)的关键因子HUPF1(human upframeshift protein 1, HUPF1)后得到明显改善[22]。(3) C9ORF72基因: 反义寡核苷酸(antisense oligonucleotide therapy, ASOs)针对C9ORF72mRNA的治疗能够缓解来源于C9ORF72突变的ALS患者诱导性多潜能干细胞分化的神经元RNA毒性。同时,ASOs能够选择性的减少C9ORF72突变的ALS患者成纤维细胞GGGGCC RNA位点而不影响整体非编码C9orf72RNA[23]。
从外源性给予神经保护因子的角度来说,包含神经营养因子在内的多个蛋白已经有治疗性潜能,并且部分已经用于ALS患者临床试验。在SOD1G93A模型鼠中,AAV介导肌肉内转染胶质源性神经营养因子(glial cell line-derived neural factor, GDNF)或者IGF-1,以及肌肉内慢病毒转染血管内皮生长因子VEGF,都观察到ALS起病时间延迟,行为学测试和运动功能改善,并且生存期延长。同时AAV定向转染IGF-1、VEGF或者G-CSF至SOD1G93A鼠中枢神经系统不同部位均能够部分改善症状并延长生存期。间充质干细胞治疗合并过表达GDNF的慢病毒转染至SOD1G93A模型鼠中时,骨骼肌肉、运动神经元和神经肌肉连接处均表现出明显的治疗作用,增加存活率。另外,SOD1G93A模型鼠中过表达人类间充质干细胞VEGF和GDNF能增加神经元和神经肌肉连接,延缓起病时间和延长生存期。
综上所述,基因治疗是目前为止最有前景的靶向治疗之一,将会给ALS患者带来福音,但同时也面临严峻的挑战: (1) ALS致病机制仍不能完全明确;(2) 有治疗性作用的分子尚缺乏有效的作用位点转入途径,同时还有类似血脑屏障等的阻碍因素阻止其转入;(3) 尚缺乏精准定位中枢神经系统特定位点和细胞的方法;(4) 神经退行性疾病的快速进展和随之而来较短的治疗时间窗;(5) 现存的ALS动物模型尚不能有效的应用于临床。因此,在ALS基因治疗道路上仍需解决这些问题。
【参考文献】
[1] DENNYS C N, ARMSTRONG J, LEVY M, et al. Chronic inhibitory effect of riluzole on trophic factor production[J]. Exp Neurol, 2015,271: 301-307.
[2] MITCHELL J, BORASIO G. Amyotrophic lateral sclerosis[J]. Lancet, 2007,369: 2031-2041.
[3] RENTON A E, CHIO A, TRAYNOR B J. State of play in amyotrophic lateral sclerosis genetics[J]. Nat Neurosci, 2014,17(1): 17-23.
[4] CHEN H J, ANAGNOSTOU G, CHAI A, et al. Characterization of the propertoes of a novel mutation in VAPB in familial amyotrophic lateral sclerosis[J]. J Biol Chem, 2010,285(51): 40266-40281.
[5] ROSS O A, RUTHERFORD N J, BAKER M, et al. Ataxin-2 repeat-length variation and neurodegeneration[J]. Hum Mol Genet, 2011,20(16): 3207-3212.
[6] MAVLYUTOV T A, EPSTEIN M L, ANDERSEN K A, et al. The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study[J]. Neuroscience, 2010,167(2): 247-255.
[7] WU C H, FALLINI C, TICOZZI N, et al. Mutations in the profiling 1 gen cause familial amyotrophic lateral sclerosis[J]. Nature, 2012,488(7412): 499-503.
[8] SAEED M, YANG Y, DENG H X, et al. Age and founder effect of SOD1 A4V mutation causing ALS[J]. Neurology, 2009,72: 634-639.
[9] YAMASHITA S, ANDO Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis[J]. Transl Neurodegener, 2015,7(24): 4-13.
[10] RADEMAKERS R, STEWART H, Dejesus-Hemandez M, et al. Fus gene mutations in familial and sporadic amyotrophic lateralsclerosis[J]. Muscle Nerve, 2010,42(2): 170-176.
[11] BUMER D, HILTON D, PAINE S M, et al. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations[J]. Neurology, 2010,75(7): 611-618.
[12] Therrien M, Dion P A, Rouleau G A. ALS: Recent developments from genetics studies[J]. Curr Neurol Neurosci Rep, 2016,16: 59-62.
[13] KATSUNO M, ADACHI H, BANNO H, et al. Transforming growth factor-β signaling in motor neuron diseases[J]. Curr Mol Med, 2011,11: 48-56.
[14] SASAKI S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis[J]. J Neuropathol Exp Neurol, 2011,70(5): 349-359.
[15] GARRITY-MOSES M E, TENG Q, LIU J, et al. Neuroprotectiveadeno-associat-ed virus Bcl-xL gene transfer in models of motor neuron disease[J]. Muscle Nerve, 2005,32: 734-744.
[16] NANOU A, HIGGINBOTTOM A, VALORI C F, et al. Viral delivery of antioxidant genes as a therapeutic strategy in experimental models of amyotrophic lateral sclerosis[J]. Mol Ther, 2013,21(8): 1486-1496.
[17] PATEL P, KRIZ J, GRAVEL M, et al. Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis[J]. Mol Ther, 2014,22(3): 498-510.
[18] YAMANAKA K, CHUN S J, BOILLEE S, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis[J]. Nat Neurosci, 2008,11: 251-253.
[19] AYERS J I, FROMHOLT S, SINYAVSKAYA O, et al. Widespread and efficient transduction of spinal cord and brain following neonatal AAV injection and potential disease modifying effect in ALS mice[J]. Mol Ther, 2015,23(1): 53-62.
[20] MILLER T M, PESTRONK A, DAVID W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study[J]. Lancet Neurol, 2013,12(5): 435-442.
[21] YAMASHITA T, CHAI H L, TERAMOTO S, et al. Rescue of amyotrophic lateral sclerosis phenotype in a mouse model by intravenous AAV9-ADAR2 delivery to motor neurons[J]. EMBO Mol Med,2013,5(11): 1710-1719.
[22] JACKSON K L, DAYTON R D, ORCHARD E A, et al. Preservation of forelimb function by UPF1 gene therapy in a rat model of TDP- 43-induced motor paralysis[J]. Gene Ther, 2015,22(1): 20-28.
[23] LAGIER-TOURENNE C, BAUGHN M, RIGO F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration[J]. Proc Natl Acad Sci USA, 2013,110(47): E4530-E4539.
ALS: Recent development from genetic and clinical study
【Abstract】 Amyotrophic lateral sclerosis (ALS) is a chronic progressive neurodegenerative disease affecting both the upper and lower motor neurons,which leads to paralysis and muscle atrophy of patients. This article reviews the relationship between different genotypes and clinical features of ALS based on to date studies.
【Key words】 amyotrophic lateral sclerosis; genotype; clinical feature; gene diagnosis; gene therapy
doi: 10.16118/j.1008-0392.2017.04.024
收稿日期: 2016-05-24
基金项目: 上海市科委重点基础项目(13JC1407103)
【中图分类号】 R746
【文献标志码】
【文章编号】 1008-0392(2017)04-0117-06