·基础研究·
【摘要】目的 研究白血病抑制因子(leukemia inhibitory factor, LIF)对肾间质纤维化的作用及其机制。方法 TGF-β和(或)LIF和(或)TSA刺激肾脏成纤维细胞(NRK-49F);TGF-β和(或)LIF刺激Stat3基因敲除细胞(MEF-Stat3-KO);用KM小鼠建立单侧输尿管梗阻(unilateral ureteral obstruction, UUO)模型,腹腔内注射LIF后收集肾脏;用Western Blot方法检测纤维化相关性蛋白(Col3和FN1)和乙酰化Stat3的表达,用RT-PCR方法检测Col3和FN1的mRNA表达,用免疫组化方法检测肾脏组织的Col3、FN1和LIFR、Stat3的沉积。结果 在NRK-49F细胞中,LIF降低TGF-β诱导的Col3和FN1的表达;在Stat3-KO细胞中,LIF对Col3和FN1的表达无作用。UUO肾脏中,LIFR和Stat3的表达较假手术组明显减少,LIF注射后Col3和FN1的沉积减少。LIF刺激NRK-49F细胞15min后,乙酰化Stat3明显增多;TSA刺激细胞后,Col3和FN1的表达降低更加明显。结论 LIF抑制体内外肾间质纤维化,依赖于Stat3,其机制可能包括乙酰化修饰。
【关键词】白血病抑制因子; 肾间质纤维化; 信号传导子及转录激活子3; 大鼠
白血病抑制因子(leukemia inhibitory factor, LIF)属白介素-6细胞因子家族,除了具有调节肿瘤生长、维持胚胎干细胞多能性等作用外[1-2],还参与调节脏器纤维化[3]。研究发现,LIF可降低心肌细胞的纤维化程度[4]。研究发现LIF可抑制血管紧张素(Angiotensin II, AngII)诱导的细胞外基质沉积,并探索了其机制可能是通过磷酸化途径[5],那么LIF对其他刺激诱导的肾间质纤维化是否也存在抑制作用,作用机制如何,本研究将进一步证明LIF对肾间质纤维化的抵抗作用,以及乙酰化修饰在其中的作用。
1.1 细胞培养
大鼠肾成纤维细胞NRK-49F由美国布朗大学庄守纲教授馈赠,小鼠胚胎成纤维细胞Stat3敲除型(MEF-Stat3-KO)由中国科学院健康所肿瘤与干细胞研究课题组秦樾教授馈赠;细胞用含10%FBS的DMEM培养液培养,刺激前0.5%FBS饥饿12~24h。TGF-β(2ng/ml)、LIF(2000U/L)、TSA(100nmol/L)刺激细胞后收集蛋白和RNA。
1.2 细胞转染
接种适当数量的细胞于培养板中,每孔中加入不含抗生素的培养基,使转染时的细胞密度能够达到30%~50%;对于每个转染样品,按Lipo 2000的说明书操作,准备siRNA转染液,6h后换正常完全培养液,24~48h后收集以进行下一步检测。
1.3 UUO模型
健康雄性清洁级昆明小鼠: 4~6周龄,约25g,购自中国科学院上海实验动物中心。体内实验分为四组: 假手术组、UUO模型组、UUO+LIF组、UUO+ 生理盐水组,每组各6只。UUO模型建立方法参见[5]。LIF给予方法: 术后第1天开始,UUO+LIF组和UUO+生理盐水组予腹腔注射LIF(每日25mg·kg-1)或相同体积生理盐水,共6d。手术后第7天处死小鼠,取肾组织供进一步实验用。
1.4 Western Blot
细胞用PBS洗后裂解;肾组织剪碎后,加裂解液,用玻璃匀浆器匀浆,离心取上清上样。聚丙烯酰胺凝胶电泳,转膜(三明治法),室温封闭1h,一抗孵育(Col3: santacruz;FN1: sigma;β-actin: millpore; Stat3、ac-Stat3: CST)4℃过夜;孵育二抗,显影,Quantity One分析灰度值,3次以上结果统计分析。
1.5 反转录-聚合酶链反应(RT-PCR)
Trizol法提取总RNA,反转录,参照TOYOBO SYBR说明书设计反应体系,进行PCR反应,取PCR产物进行琼脂糖凝胶电泳,溴化乙锭染色,凝胶成象仪成象。
1.6 免疫组织化学
肾脏组织经固定、脱水、透明及浸蜡处理后切为厚度为3~5mm的石蜡切片。切片经脱蜡水化、抗原修复、去内源性过氧化酶、封闭、孵育一抗(Col3、FN1: abcam;Stat3: CST),生物素标记二抗孵育,辣根过氧化物标记的三抗孵育,DAB显色,苏木精复染,脱水及透明,中性树胶封片,光镜观察。
1.7 数据分析
所有数据以![]() ±s表示,利用SPSS 17.0软件,采用单因素ANOVA和t检验分析。P<0.05具有统计学意义。
±s表示,利用SPSS 17.0软件,采用单因素ANOVA和t检验分析。P<0.05具有统计学意义。
2.1 LIF抑制TGF-β诱导的细胞外基质蛋白的表达
细胞外基质(extracellular matrix, ECM)沉积被认为是肾间质纤维化的主要病理改变,选用产生ECM的最主要细胞-大鼠成纤维细胞系-NRK-49F,选取ECM的主要成分—3型胶原蛋白(Collagen 3, Col3)和纤连蛋白(Fibronectin, FN1)作为研究指标。转化生长因子(transforming growth factor-β, TGF-β)是公认的促纤维化因子[6]。在NRK-49F细胞中,TGF-β促进Col3的蛋白表达,而同时给予LIF刺激后,Col3的蛋白表达受到抑制(图1A、1B);FN1(图1A、1B)的蛋白表达呈现相同的趋势。TGF-β明显上调FN1在mRNA水平的表达,而LIF可抑制这种作用(图1C、1D)。
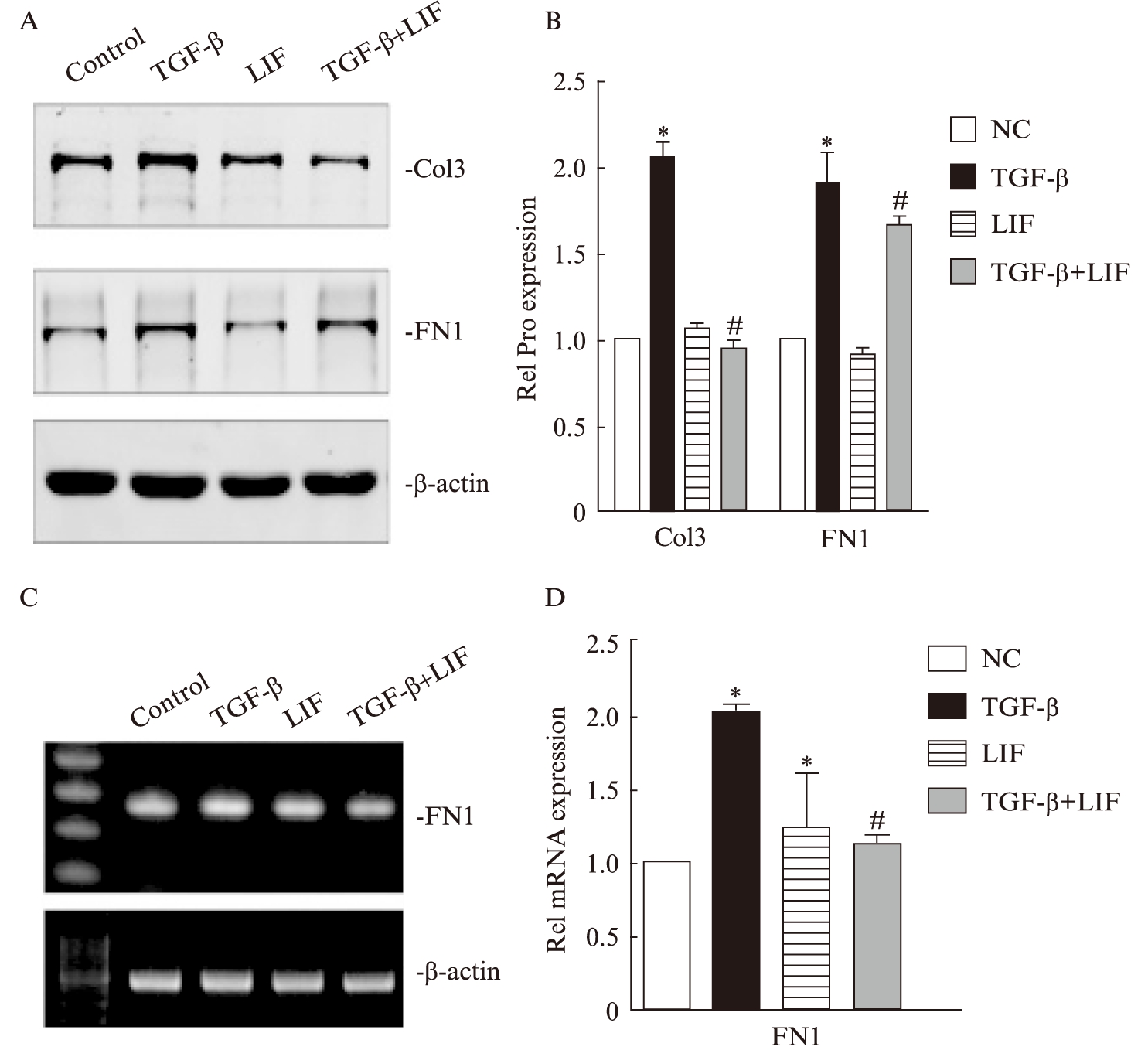
图1 LIF抑制TGF-β诱导的细胞外基质蛋白的表达
Fig.1 LIF inhibited of TGF-β induced expression of extracellular matrix proteins
予NRK-49F细胞TGF-β和(或)LIF刺激后Col3和FN1(A)的蛋白表达,以及灰度统计值(B);TGF-β和(或)LIF刺激后FN1的mRNA水平的表达(C)以及半定量统计值(B)。n≥3,*P<0.05 vs. 对照组,#P<0.05 vs. TGF-β刺激组
2.2 UUO模型中细胞外基质蛋白增加,LIF受体减少
选择单侧输尿管梗阻(Unilateral ureteral obstruction, UUO)的小鼠作为体内肾间质纤维化的模型。免疫组化染色可见Col3和FN1的沉积明显增加(图2A),同时,LIF的受体LIFR(leukemia inhibitory factor receptor)和其下游转录因子Stat3的表达明显减少(图2B)。
2.3 LIF抑制小鼠UUO模型的肾间质纤维化
为了研究LIF对肾间质纤维化的作用,予UUO小鼠腹腔注射LIF。可见: LIF干预明显下调UUO导致的Col3和FN1(图3A)在肾小管间质中的沉积。而LIF注射对其他脏器(心脏、肝脏、脾脏、肺脏)的形态无明显影响(图3B)。这些结果说明LIF在体内外均可抑制肾间质纤维化。
2.4 LIF对肾间质纤维化的作用依赖核转录因子Stat3
研究发现LIF对AngII诱导的肾间质纤维化的抵抗作用依赖于其下游核转录因子Stat3(signal conduction and activation of transcription 3),使用TGF-β刺激,进一步确定Stat3在LIF对肾间质纤维化中的作用。首先,在Stat3敲除的MEF细胞(Stat3-/-)中观察到TGF-β仍可诱导Col3和FN1的表达,而LIF的抑制作用消失(图4A、B)。进一步,在NRK-49F细胞中转染si-Stat3,观察到Stat3的表达明显下调(图4C、D),再给予细胞TGF-β和(或)LIF刺激后,如同MEF细胞中观察到的一样,TGF-β对Col3和FN1的诱导作用存在,而LIF的抑制作用则消失了(图4E、F)。这部分结果进一步证明了Stat3在LIF对抗肾间质纤维化作用的信号通路中的关键作用。
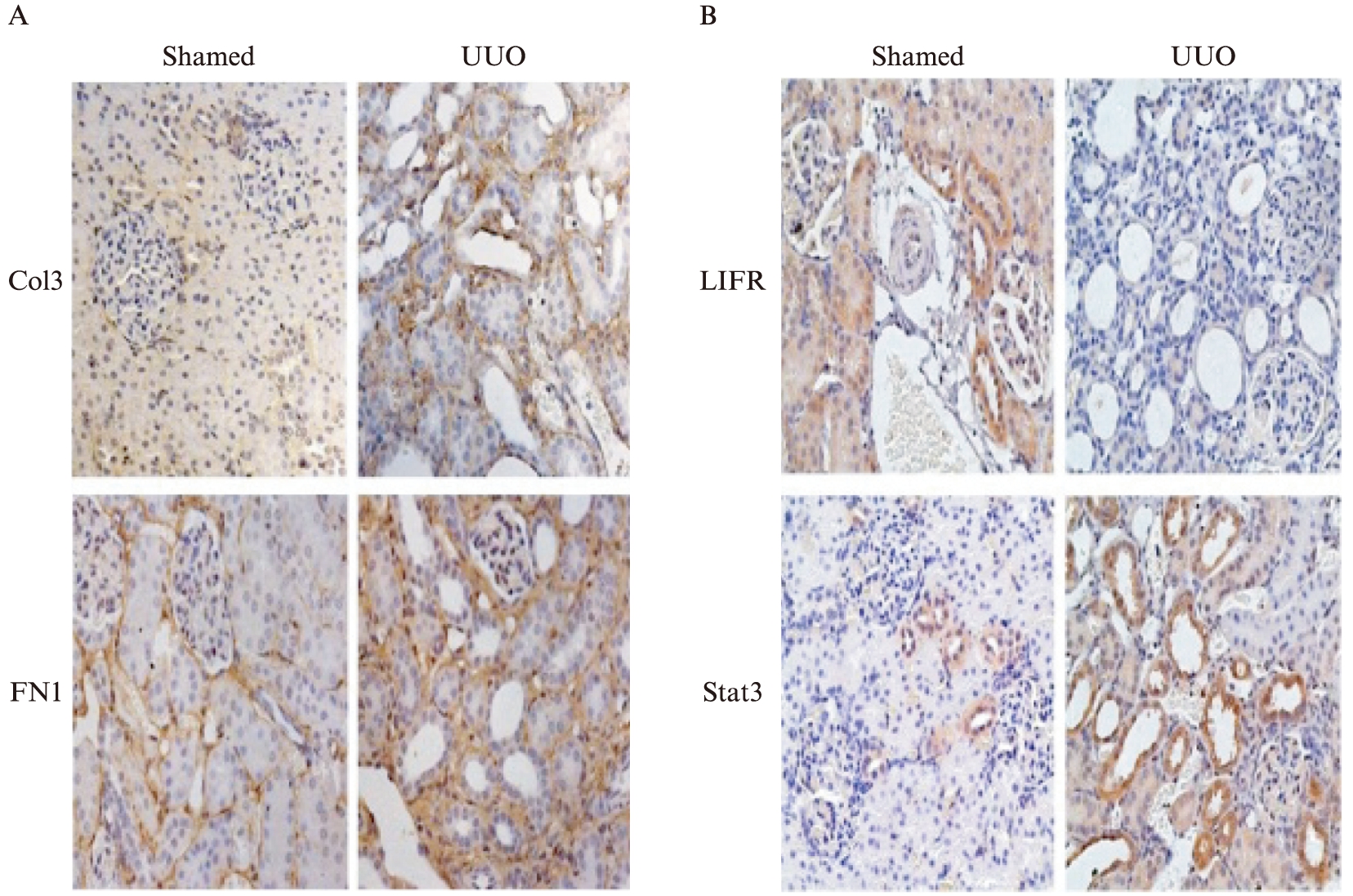
图2 UUO模型中细胞外基质蛋白、LIF受体和Stat3表达情况
Fig.2 The expression of extracellular matrix proteins, LIF receptor and Stat3 in UUO model
A: 特异性抗Col3和FN1免疫组化染色(400x);B: 特异性抗LIFR和Stat3免疫组化染色(400x)
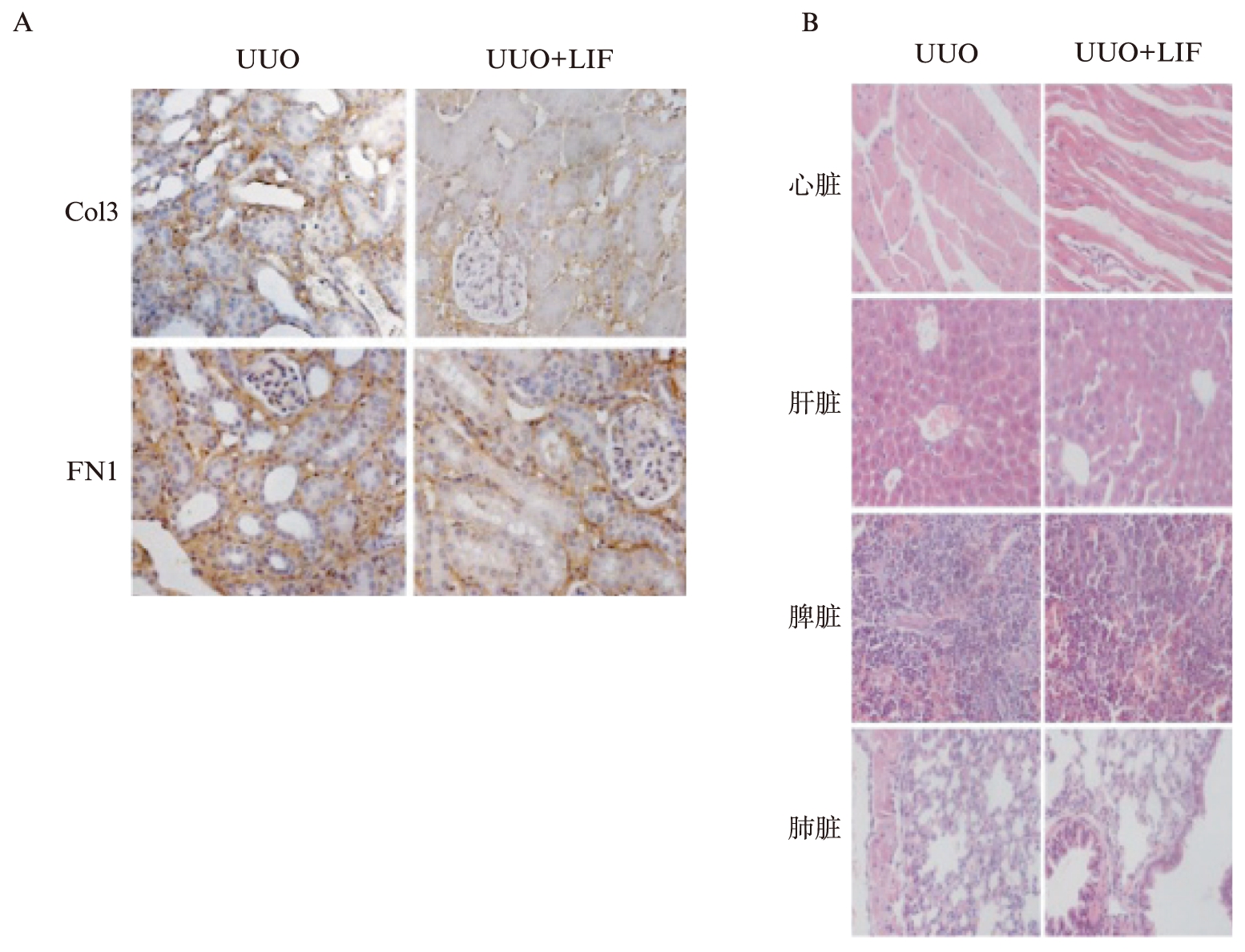
图3 LIF对UUO模型的作用
Fig.3 The effect of LIF in UUO model
A: 特异性抗Col3和FN1的免疫组化染色;B: LIF处理前后UUO模型的心脏、肝脏、脾脏和肺脏的形态学表现(H-E)
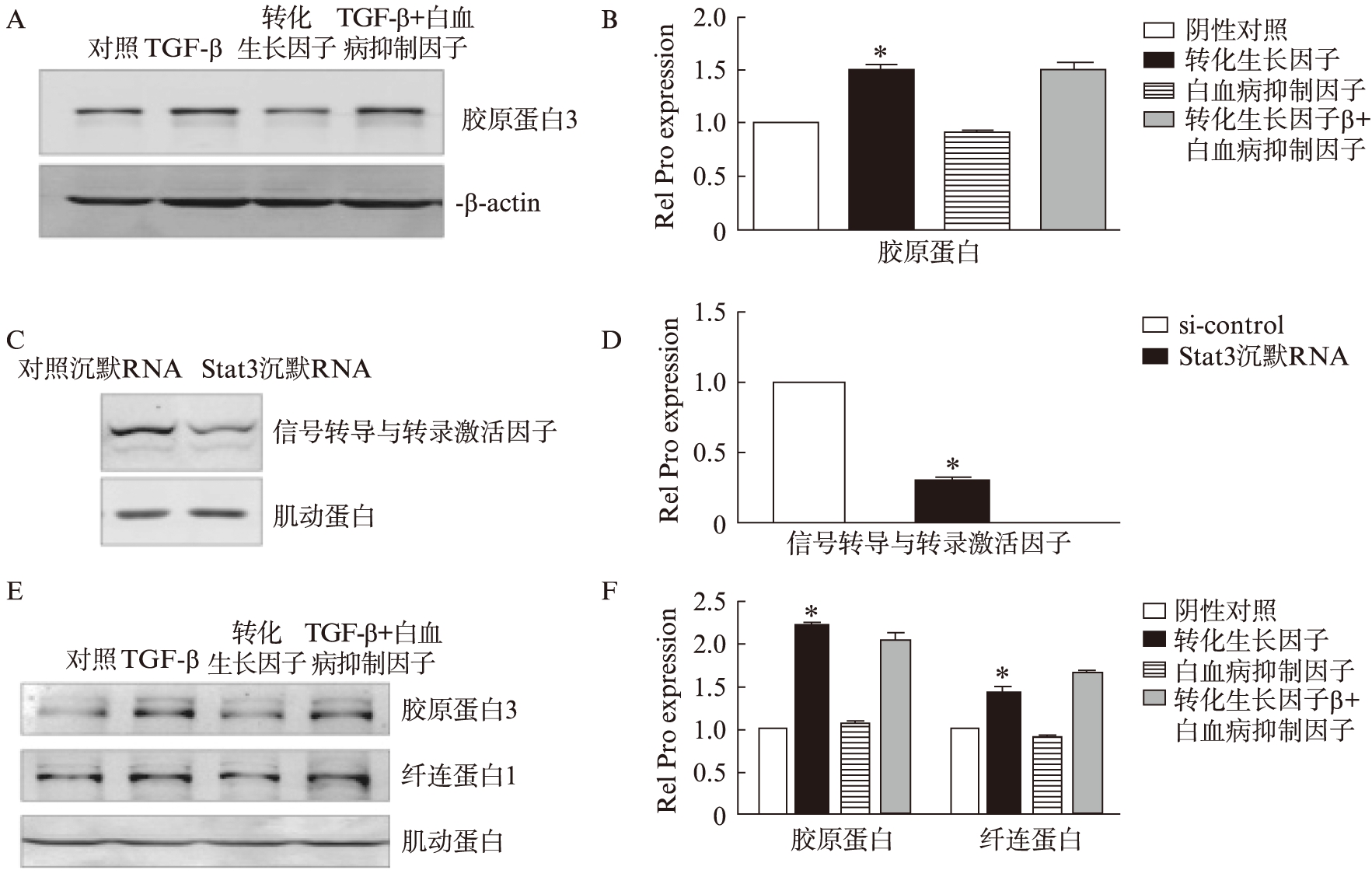
图4 LIF对肾间质纤维化的作用依赖核转录因子Stat3
Fig.4 The effect of LIF on renal interstitial fibrosis was dependent on the nuclear transcription factors-Stat3
Stat3基因敲除的MEF细胞经TGF-β和/或LIF刺激后,Col3的蛋白表达水平(A),以及灰度值统计分析(B)。NRK-49F细胞转染对照si-RNA(si-control)或Stat3的siRNA(si-Stat3)后检测Stat3的蛋白表达(C),并做灰度值统计分析(D)。NRK-49F细胞转染si-Stat3后,予TGF-β和/或LIF刺激后,Col3和FN1的蛋白表达水平(E),以及灰度值统计分析(F)。 n≥3,*P<0.05 vs. 对照组
2.5 LIF诱导Stat3乙酰化
除了磷酸化修饰,乙酰化修饰在信号转导通路中也发挥重要的作用。观察到: LIF刺激细胞后,核转录因子Stat3发生乙酰化修饰(图3): 在NRK-49F细胞中,给予LIF刺激5min,15min,30min,60min,120min 后,观察到在LIF刺激15min 和30min时出现明显的乙酰化Stat3条带,60min和120min时稍微减弱,但仍较无刺激组条带明显(图5)。
2.6 乙酰化修饰参与LIF抑制肾间质纤维化作用
组蛋白乙酰化转移酶(histone acetylation transferase, HATs)和组蛋白去乙酰化酶(histone acetylation enzyme, HDACs)之间的平衡调节蛋白的乙酰化状态。使用HDAC特异性抑制剂-TSA(Trichostation A)刺激细胞后发现: TSA+TGF- β-+LIF组的Col3和FN1的表达较TGF-β-+LIF组的更加减少(图6),说明TSA可加强LIF对TGF-β-诱导的ECM的抑制作用。
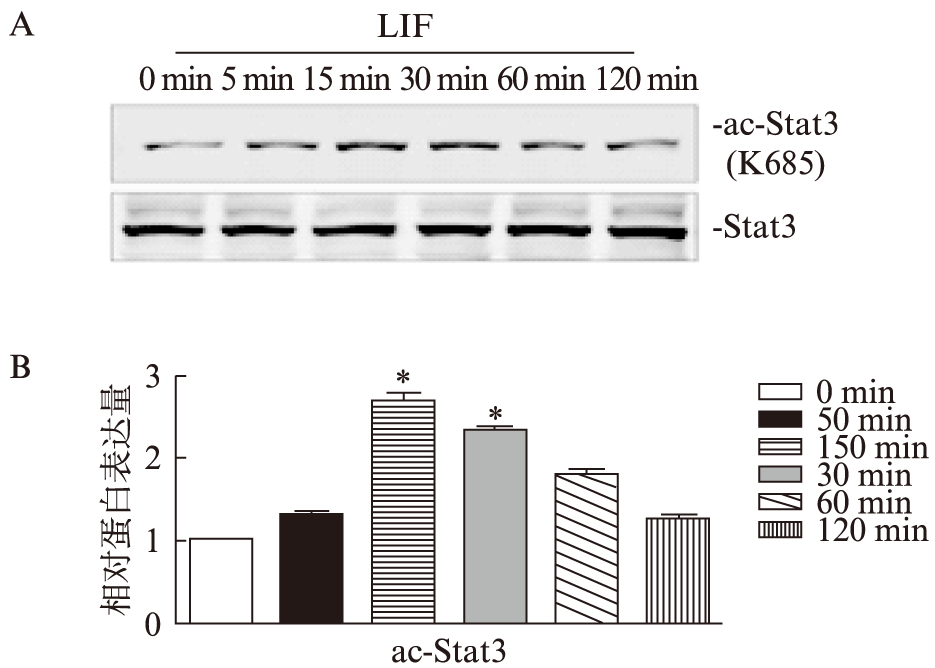
图5 LIF诱导Stat3乙酰化
Fig.5 LIF induced the acetylation of Stat3
NRK-49F细胞中,LIF刺激不同时间后Stat3的685位点赖氨酸的乙酰 化化变化(A)以及灰度分析(B),n≥3,*P<0.05 vs. 0min 组
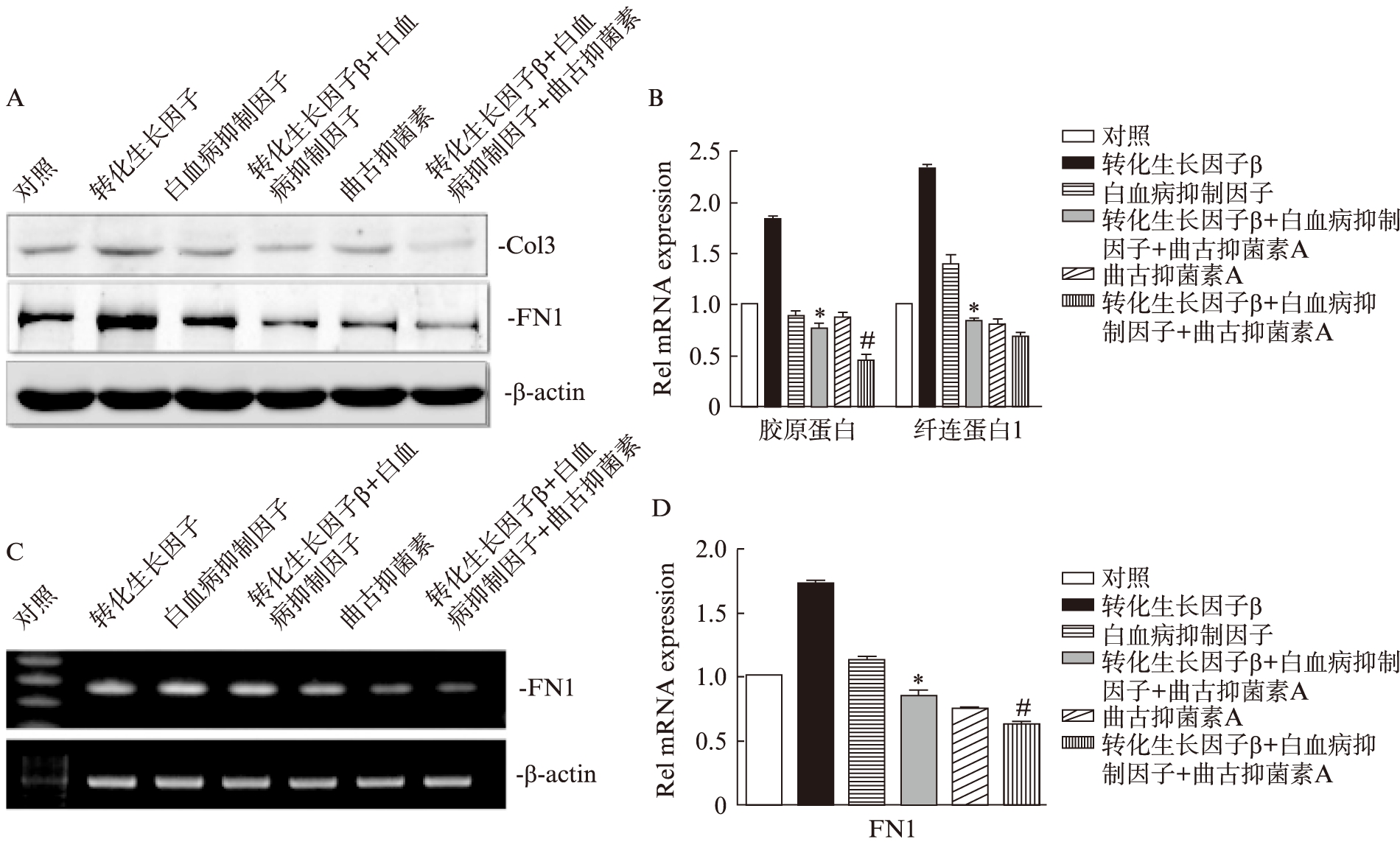
图6 TSA加强LIF对TGF-β诱导的细胞外基质表达的抑制作用
Fig.6 TSA strengthened the inhibition of LIF on TGF-β-induced of extracellular matrix expression
予NRK-49F细胞TGF-β和(或)LIF刺激后,或同时加TSA刺激,检测Col3和FN1的蛋白表达水平(A)以及FN1的mRNA表达(C),并做灰度值统计分析(B、D). TGF-β刺激组*P<0.05,TGF-β+LIF刺激组#P<0.05
LIF具有维持胚胎干细胞的多能性、促进子宫内膜蜕膜化、调节肿瘤生长等多种生物功能[7-8]。研究发现LIF可调节脏器纤维化。LIF可促进心肌成纤维细胞增殖,明显减少胶原蛋白表达,同时抑制成纤维细胞转化为肌成纤维细胞[9]。实验观察到LIF抑制AngII诱导的ECM表达,本实验选择促纤维化因子TGF-β处理细胞,观察到LIF对TGF-β导致的ECM表达亦存在抑制作用,说明LIF对肾间质纤维化的作用是广泛的,而非AngII特异性的。体内实验方面,在UUO模型的肾脏组织中观察到LIFR表达明显增多,提示LIF通过其受体LIFR参与调节肾间质纤维化的形成;进一步,UUO小鼠注射LIF后,肾间质纤维化的程度减轻,而不影响其他重要脏器的形态。
之前的实验[5]认为LIF对肾间质纤维化的作用主要通过Stat3,但是Stat3为AngII和LIF的共同下游核转录因子,不能排除LIF是通过AngII而作用于Stat3的,选择下游为主要为非Stat3的TGF-β进行实验。本实验中,Stat3表达缺失或下调后,TGF-β的作用仍存在,而LIF的作用消失,这更加证明LIF对抗肾间质纤维化的作用是依赖Stat3的。
近年来,文献报道称,乙酰化修饰在肾间质纤维化形成过程中占有重要的地位。在较早以前,乙酰化修饰特指组蛋白的乙酰化修饰,属于表观遗传修饰的一种,但是越来越多的研究表明,除了组蛋白外,其他蛋白质也可以发生乙酰化修饰。Eugene Chin教授等[10-11]首次发现报道了: 细胞膜的细胞因子受体与细胞浆内STATs,不仅发生磷酸化,还发生乙酰化修饰。发现Stat3 685位点赖氨酸是重要的乙酰化位点,在调节肿瘤生长中具有重要的作用。
本实验观察到, LIF刺激可诱导其下游Stat3发生乙酰化修饰;在给予TGF-β+LIF刺激的同时给予TSA刺激后观察到ECM蛋白水平下降较TGF-β+ LIF组更加明显。TSA是HDAC的抑制剂,可增加细胞乙酰化修饰的水平。TSA增加了LIF抗肾间质纤维化的作用,即提示乙酰化修饰参与LIF的作用。文献报道,TSA可抑制肺脏、肝脏、心脏等的纤维化[12-13],说明乙酰化修饰对纤维化的重要作用。结果与文献报道相符合: HDAC抑制剂可明显减轻肾间质纤维化的程度[14-15]。
综上所述,本实验观察到LIF抑制TGF-β诱导的ECM蛋白表达,LIF抑制UUO模型中的ECM沉积;Stat3表达下调后,LIF的作用消失;LIF诱导Stat3发生乙酰化修饰,而乙酰化水平的上调增强LIF的作用。本实验进一步证明了LIF对肾间质纤维化的抑制作用,至少部分是通过乙酰化Stat3完成的。
【参考文献】
[1] 李明堂,付玉,闫东君. LIF: 一种多功能的细胞因子[J].中国实验诊断学,2007,11(7): 987-990.
[2] Suman P, Malhotra SS, Gupta SK. LIF-STAT signaling and trophoblast biology[J]. J Stat, 2013,2(4): e25155.
[3] Berry MF, Pirolli TJ, Jayasankar V, et al. Targeted overexpression of leukemia inhibitory factor to preserve myocardium in a rat model of postinfarction heart failure[J].J Thorac Cardiovasc Surg, 2004,128(6): 866-875.
[4] Masato K, Toshio N, Toshinao T, et al. Leukemia inhibitory factor enhances endogenous cardiomyocyte regeneration after myocardial infarction[J]. Plos One, 2016,11(5): e0156562.
[5] Yu Y, Wang Y, Niu Y, et al. Leukemia inhibitory factor attenuates renal fibrosis through Stat3-miR-29c[J]. Am J Physiol Renal Physiol, 2015,309(7): F595-603.
[6] 芮炜玮,易祥华.转化生长因子β1及相关细胞因子在肺纤维化发生中的作用[J],同济大学学报: 医学版,2010,31(3): 133-136.
[7] Salleh N, Giribabu N. Leukemia inhibitory factor: roles in embryo implantation and in nonhormonal-contraception[J]. The Scientific World J, 2014,2014: 201514.
[8] Yue X, Wu L, Hu W. The regulation of leukemia inhibitory factor[J]. Cancer Cell Microenviron, 2015,2(3): e877.
[9] Wang F, Trial J, Diwan A, et al. Regulation of cardic fibroblast cellular function by leukemia inhibitory factor[J]. J Mol Cell Cardiol, 2002,34(10): 1309-1316.
[10] Tang X, Gao JS, Guan YJ, et al. Acetylation-dependent signal transduction for type I interferon receptor[J]. Cell, 2007,131, 93-105.
[11] Yuan ZL, Guan YJ, Chatterjee D, et al. Stat3 dimerization regulated by reversible acetylation of a single lysine residue[J].Science, 2005,307(5707): 269-273.
[12] Sanders YY, Tollefsbol TO, Varisco BM, et al. Epigenetic regulation of thy-1 by histone deacetylase inhibitor in rat lung fibroblasts[J].Am J Respir Cell Mol Biol, 2011,45(1): 16-23.
[13] Dai Q, Liu J, Du Y, et al. Histone deacetylase inhibitors attenuate P-aIgA1-induced cell proliferation and extracellular matrix synthesis in human renal mesangial cells in vitro[J]. Acta Pharmacol Sin, 2016,37(2): 228-234.
[14] Yoshikawa M, Hishikawa K, Marumo T, et al. Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-beta1 in human renal epithelial cells[J]. J Am Soc Nephrol, 2007,18: 58-65.
[15] Imai N, Hishikawa K, Marumo T, et al. Inhibition of histone deacetylase activates side population cells in kidney and partially reverses chronic renal injury[J]. Stem Cells, 2007,25: 2469-2475.
Inhibitory of renal interstitial fibrosis by acetylation of STAT3 with white cell inhibitor
【Abstract】Objective To investigate the effect of leukemia inhibitory factor(LIF) on renal interstitial fibrosis and its mechanism. Methods Renal fibroblasts(NRK-49F) were stimulated with TGF-beta and/or LIF, and/or TSA. Stat3 gene knockout cells(MEF-Stat3-KO) were stimulated with TGF-beta and/or LIF. Unilateral ureteral obstruction(UUO) was induced in KM mice and kidneys were collected after intraperitoneal injection of LIF. Western Blot method was used to detect protein expression of collagen 3, fibronectin and acetylated Stat3. RT-PCR method was used to detect mRNA expression of collagen 3 and Fibronectin, and immunohistochemical method was used to detect protein deposition of collagen 3, fibronectin, LIFR and Stat3 in kidney tissue. Results In NRK- 49F cells, LIF inhibited TGF-beta-induced Col3 and FN1 expression. In Stat3-KO cells, LIF had no effect on the expression of Col3 and FN1. In UUO kidney, the expression of LIFR and Stat3 decreased significantly compared with that in the control group; after injection of LIF, the deposition of Col3 and FN1 reduced. After stimulation with LIF for 15min, acetylated Stat3 in NRK-49F cells increased significantly. TSA stimulation reduced the expression of Col3 and FN1 more markedly. Conclusion LIF can inhibit renal interstitial fibrosis bothin vitro and in vivo, which may be associated with the acetylation of Stat3.
【Key words】Leukemia inhibitory factor; renal interstitial fibrosis; signal conduction and activation of transcription 3; rat
doi:10.16118/j.1008-0392.2016.06.009
收稿日期:2016-08-05
基金项目:国家自然科学基金(81370790、81600523)
【中图分类号】R 692.5
【文献标志码】A
【文章编号】1008-0392(2016)06-0047-07