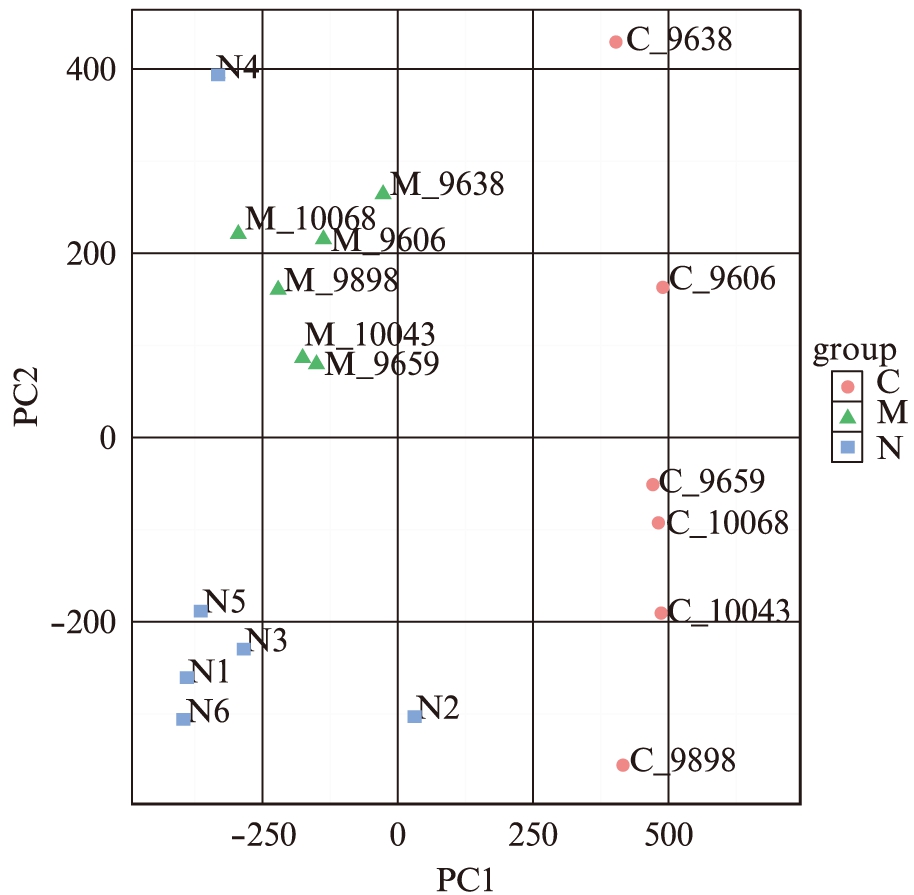
图1 M,N和C三组样品进行二维PCA分析分析结果
Fig.1 Three M, N and C 1 groups of samples for two-dimensional PCA analysis of the results
·基础研究·
【摘要】目的 子宫内膜异位症(endometriosis, EM)是妇科的多发病和常见病,发病率逐年增加。但内异症的发病机制至今并不明确。 EM类似恶性肿瘤的生物学行为可能是EM重要的发病机制之一,而恶性病变的发生、发展是以抑癌基因和癌基因遗传变异的积累为标志的,尤其是抑癌基因的异常甲基化。方法 抑癌基因非甲基化的CpG岛异常甲基化与人类恶性肿瘤的发生、转移密切相关。本实验采用成组病例对照方法,选取以手术病理确诊为卵巢型EM的汉族妇女6例为研究对象,选取患者的在位内膜组织标本和异位内膜组织标本各6例,选择同期住院患者中族别、年龄、文化程度等构成比相似的非EM的汉族妇女6例为对照组,选取患者的正常在位内膜组织标本6例。采用美国illumina公司生产的illumina Human Methylation450K Beadchip全基因组甲基化芯片进行全基因组甲基化研究,通过对全基因组甲基化芯片的总共48万个甲基化位点进行综合生物信息学分析。结果 通过对EM患者原位和异位内膜组织样品的全基因DNA甲基化修饰进行检测,并进行聚类分析、主成分分析、GO功能注释和KEGG信号通路分析,发现在EM患者的全基因甲基化修饰水平发生了显著变化。对差异甲基化的基因进行GO功能注释,结果发现富集的GO条目主要集中于免疫过程的抗原呈递和干扰素Y的相关通路,分子功能主要集中在协同转运、负离子跨膜转运等活性,而KEGG通路分析发现,1型糖尿病,自身免疫性甲状腺疾病相关信号通路被显著富集。其中,富集程度最高的通路是focal adhesion通路,参与其中的蛋白分子包括ECM-受体互作,细胞因子-受体互作,这些结果为揭示新的EM的发病分子机制提供了参考。结论 该实验为EM病因学研究提供新的理论依据,从而为EM分子生物学发病机制的研究提供新思路。
【关键词】子宫内膜; 甲基化; GO分析; KEGG
为了深入研究子宫内膜异位症(endometriosis, EM)发生中基因表达的甲基化调控,探索子宫内膜异位症患者中潜在的DNA甲基化药物作用靶点,本项目拟采用成组病例对照方法,以手术病理确诊为卵巢型EM的汉族妇女为研究对象,选择同期住院患者中族别、年龄、文化程度等构成比相似的非EM的汉族妇女为对照组,选其中EM患者的在位内膜组织标本和异位内膜组织标本,以及汉族非EM患者的在位内膜组织标本。采用美国illumina公司生产的illumina Human Methyla-tion450K Beadchip全基因组甲基化芯片进行全基因组甲基化研究。(1) 通过对全基因组甲基化芯片的总共48万个甲基化位点进行综合生物信息学分析,初步筛查出和EM发生密切相关的基因或甲基化区域;(2) 可通过最终疾病预测模型的构建,初步确定对EM最有贡献的甲基化区域或基因;(3) 为EM病因学研究提供新的理论依据,从而为EM分子生物学发病机制的研究提供新思路[1-2]。
1.1 一般资料
选取调查病例组汉族6名;对照组汉族6名,所有患者术前3个月均未接受放疗、化疗或激素治疗,所有诊断均经病理学检查证实。
病例组: 选取2013年6月至2015年6月期间在同济大学附属东方医院住院的患者行腹腔镜或开腹手术,术后病理为明确诊断为卵巢型EM的汉族患者各6例作为病例组,年龄20~48岁,分别取典型异位内膜6份(M),并分别刮取在位子宫内膜6份(C)。
对照组: 另选取同期住院的患良性病变需行妇科手术,且均需排除患有EM的 汉族患者6例作为对照组,分别刮取正常子宫内膜6份(N),这些患者的族别、年龄、文化程度、经济收入、BMI值等与病例组的构成比相似、相匹配,与病例组相比无统计学差异[3-4]。
1.2 实验方法
采用美国illumina公司生产的illumina Human Methylation450K Beadchip全基因组甲基化芯片进行全基因组甲基化检测。illumina全基因组DNA甲基化芯片是采用单碱基延伸原理,针对每个甲基化位点设计两种探针(甲基化探针和非甲基化探针)来检测,重亚硫酸盐处理后的gDNA与每种探针互补杂交,通过识别“C”和“T”完成探针的末位延伸以及甲基化的判定,甲基化程度通过比对两种探针(甲基化探针和非甲基化探针)的荧光信号强度来计算。使用该技术可以准确的检测基因组上任何类型的甲基化位点,包括CpG位点及非CpG的甲基化位点(如CA,CT)[5-6]。
1.3 数据分析
生物信息学分析,统计学处理数据分析项目如下。(1) DSAM-01: DNA甲基化芯片原始数据的预处理;(2) DSAM-02: DNA甲基化芯片相关性与重现率(重复率/相关性)分析;(3) DSAM-03: DNA甲基化芯片的处理组-对照组相关性评估;(4) DSAM-04: 甲基化/非甲基化位点(基因)Fisher和Pearson卡方统计检验;(5) DSAM-05: 样本组间的甲基化趋势差异分析;(6) DSAM-06: 样本组间的甲基化趋势(非监督/监督式)聚类分析;(7) DSAM-07: 基于甲基化谱构建疾病预后、诊断预测模型;(8) DSAM-08: 差异化甲基化位点相关上下游基因的信号通路与生物途径(Pathway)分析;(9) DSAM-09: 差异化甲基化位点相关上下游基因的生物学功能本体注释(GO)。统计软件采用SPSS 17.0[7-8]。
2.1 全基因组甲基化芯片检测子宫内膜异位中基因甲基化水平变化
选取经过病理诊断为子宫内膜异位的汉族妇女作为入组研究对象,并选择同样比例种族、年龄和教育背景非子宫内膜异位的汉族妇女作为对照组。通过Illumina公司的人450K全基因组DNA甲基化芯片对原位子宫内膜组织样品和异位子宫内膜组织样品中的480000个甲基化位点的甲基化修饰状况进行检测,以分析两组样品之间差异的甲基化修饰情况。
2.2 主成分分析
在分析子宫内膜异位组和对照组的差异甲基化的位点后,通过主成分分析对三组样品中的基因进行分析。在经过数据标准化分析后,对M,N和C三组样品进行二维PCA分析分析结果,见图1。三组样品得到清晰区分,表明EM组样本中基因的甲基化谱与对照和非EM组样品组差异明显,这一结果进一步说明DNA的甲基化修饰在EM发病过程中的重要作用。 集中分析EM和非EM组样本中基因的甲基化差异。通过分析N和M组中的甲基化差异,鉴定了差异的甲基化位点和基因。其中有95个差异的甲基化位点,与非EM组相比,有67个位点被显著高甲基化,有28个位点被显著低甲基化[9]。
2.3 GO功能分析
对差异甲基化的基因进行GO富集分析,以分析M和N组之间的差异基因所参与的功能。分析结果,见图2,在生物过程中,包括通过经由MHC Ⅱ的抗原处理和呈递,干扰素γ相关信号通路等均被显著富集,这些结果表明,免疫系统中与抗原处理呈递有关的分子可能参与EM的发生过程。另外,对细胞组分进行的分析结果表明,分布于内质网膜、内吞囊泡膜上的分子被显著富集。在分子水平,最为显著的功能主要集中在阴离子跨膜转运等活性、D4多巴胺受体结合等通路,这暗示细胞膜系统可能是EM的关键分子。
图1 M,N和C三组样品进行二维PCA分析分析结果
Fig.1 Three M, N and C 1 groups of samples for two-dimensional PCA analysis of the results
2.4 KEGG 信号通路分析
为了进一步研究EM和非EM组差异甲基化的基因所参与的信号通路,对这些基因的进行KEGG信号通路分析。根据P<0.05和 FDR<0.05的标准,挑选了最为显著的20条主要的信号通路,见图3。这些关键的信号通路包括1型糖尿病,自身免疫胸腺疾病等通路被显著富集,这些结果进一步说明EM发病过程中免疫系统的变化,这一结果也与GO分析的结果相对应。
2.5 M和C组之间信号通路分析
在分析了EM和非EM组差异甲基化的基因所参与的信号通路后,进一步对M和C组中差异甲基化的基因所参与的信号通路进行分析,并鉴定到了一条最为显著的信号通路: focal adhesion。在参与的信号通路总,包括ECM受体作用信号通路、细胞因子受体作用信号通路等被显著富集,见图4。而其中一个关键蛋白FAK在两条通路中都被显著富集,该结果提示FAK可能作为关键的调控异常甲基化的分子参与EM发病过程。
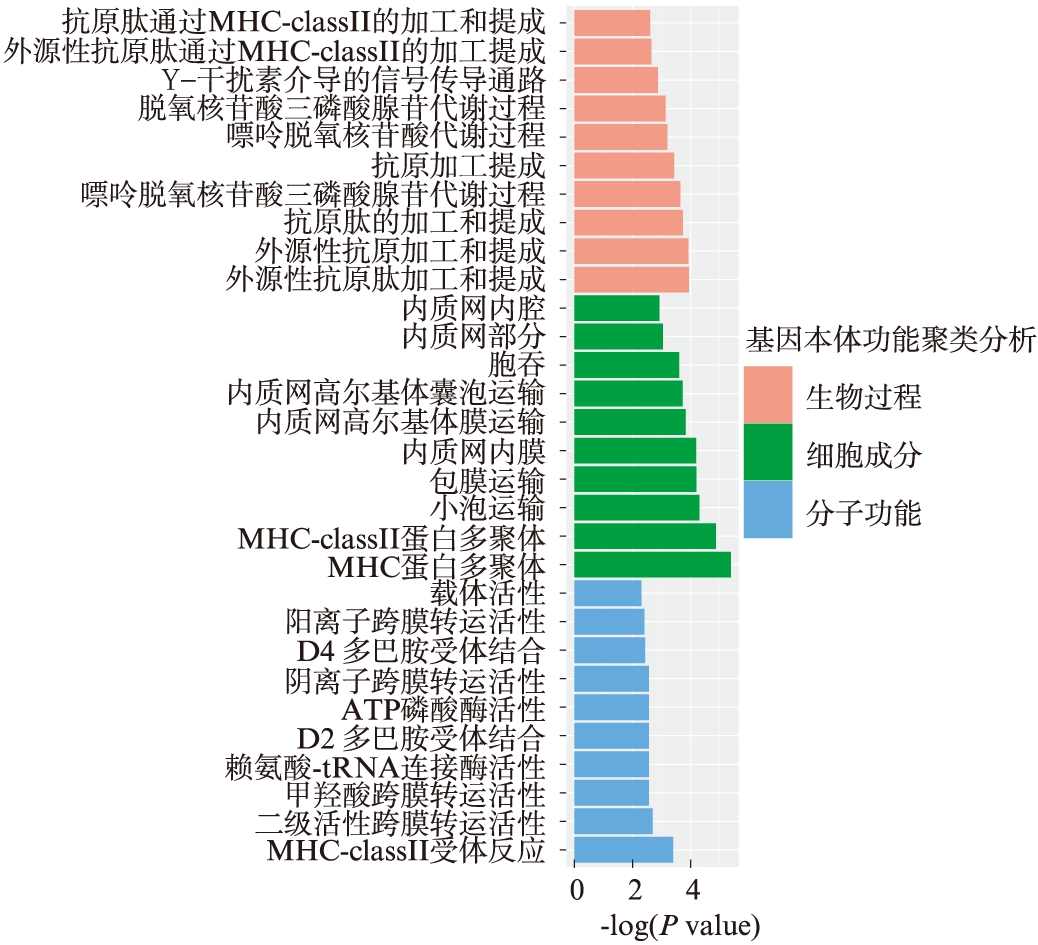
图2 分析M和N组之间的差异基因所参与的功能
Fig.2 Analysis of the function of genes involved in the differences between M and N groups
自上而下分为: 生物过程、细胞成分、分子功能

图3 KEGG通路分析显示EM中差异甲基化基因与不同疾病相关
Fig.3 KEGG pathway analysis showed that the differentially methylated genes in EM related to various diseases
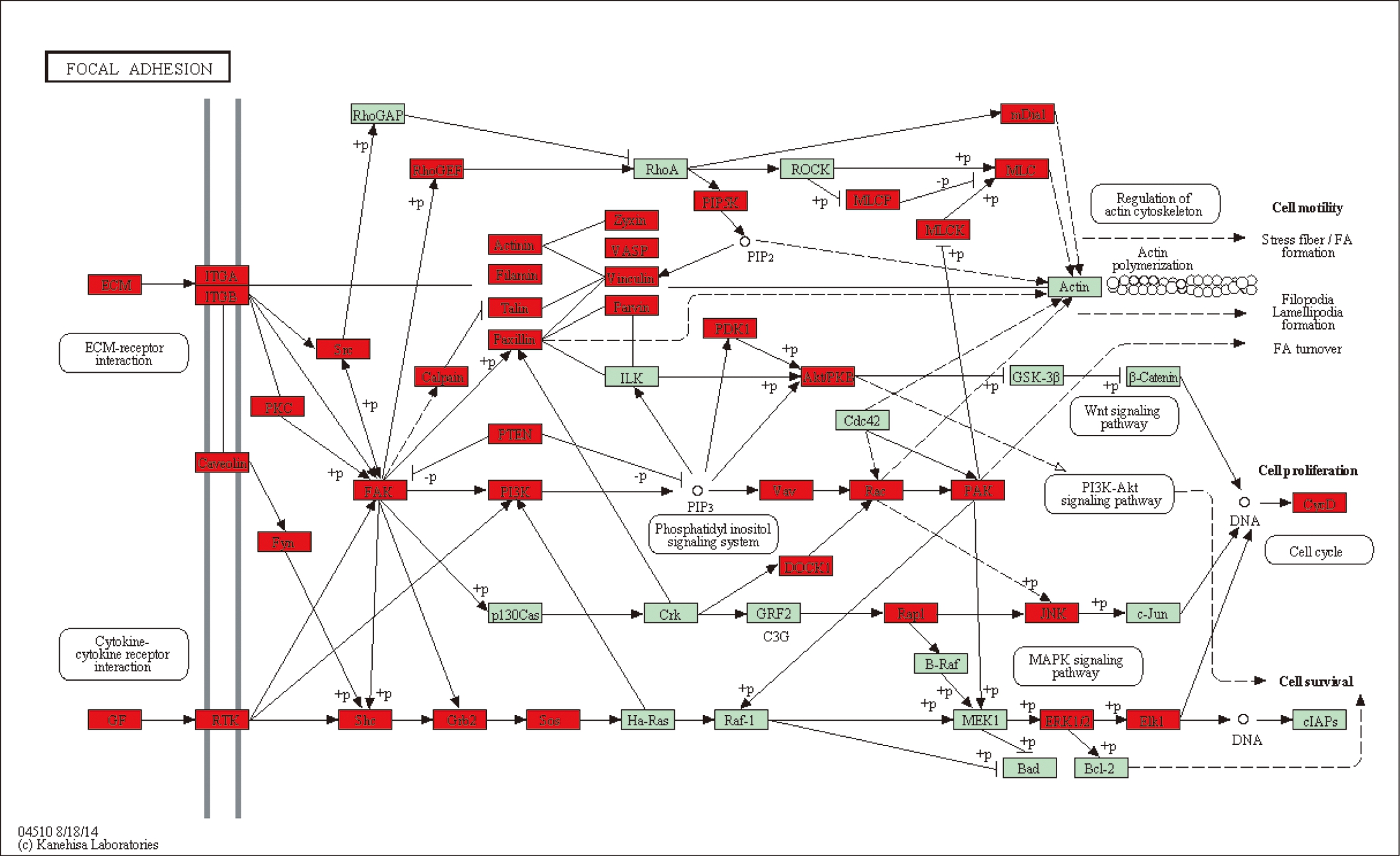
图4 在M组和C组中最明显改变的信号通路
Fig.4 The most significantly changed pathways between M and C groups
通过对EM患者原位和异位内膜组织样品的全基因DNA甲基化修饰进行检测,并进行聚类分析、主成分分析、GO功能注释和KEGG信号通路分析,发现在EM患者的全基因甲基化修饰水平发生了显著变化。对差异甲基化的基因进行GO功能注释,结果发现富集的GO条目主要集中于免疫过程的抗原呈递和IFNγ,分子功能主要集中在协同转运、负离子跨膜转运等活性,而KEGG通路分析发现,1型糖尿病,自身免疫性甲状腺疾病相关信号通路被显著富集。其中,富集程度最高的通路是focal adhesion通路,参与其中的蛋白分子包括ECM-受体互作,细胞因子-受体互作,这些结果为揭示新的EM的发病分子机制提供了参考。
在本研究结果中,发现与非EM患者相比,EM患者的整体甲基化修饰水平发生了显著变化,这提示由甲基化修饰所调控的基因表达可能参与了EM的发病,而干预改过程则有助于改善EM进展。Wu等[10]测定了EM患者中HOXA10基因的表达水平,并推测HOXA10启动子区域的异常甲基化可能是EM发生发展的重要原因[11-12]。临床干预EM的常用方式是手术和药物,然而药物治疗只能暂时缓解病症,而不能彻底根除疾病,并且术后有极高的复发率[13-14]。同时,Kim等[15]和Lee等[16]研究团队分别在狒狒和小鼠的EM动物模型中发现,EM恶性癌变的一个主要原因是HOXA10基因启动子F1区的高甲基化修饰。Eun等[17]和Lu等[18]提出HOX基因可以作为子宫内膜容受性的标志物,并通过基因治疗的方式抑制了HOX基因的甲基化,从而提高了HOX的表达水平,最后提高了怀孕率。在EM细胞模型中,基因甲基化的修饰改变抑制了炎症因子的分泌[19],并降低了EM的发病率,实验结果中,发现抗原递呈、炎症因子分泌等免疫过程被显著富集,这些结果说明DNA的异常甲基化通过调控关键分子和通路的表达在调控EM的发病过程中起到关键调控作用。
近期,有研究表明表观层面的遗传修饰,特别是DNA甲基化介导的表达异常在EM发病过程中起到重要的调节作用。Martini等发现,EM 患者中DNA损伤修复相关基因hMLH1和 p16基因CpG岛的异常甲基化修饰导致对应基因的表达缺失。由于hMLH1的缺失可能导致DNA复制过程中的间接错配,因此微卫星片段的长度和稳定性会发生变化,最终导致肿瘤发生[8]。因此,DNA甲基化的模式与EM疾病的发生和预后显著相关,并能作为EM恶化癌变的预测因子。从全基因组整体甲基化修饰的角度,对EM和非EM患者基因的甲基化修饰状态进行检测,并找到了两组中差异最显著的基因,并为EM的靶向治疗提供了新的角度和理论基础。
从表观遗传的角度来看,DNA甲基化修饰的异常解释了EM发病的分子机制,而这些异常甲基化可能成为EM患者一系列并发症的主要机制。EM发病中的表观遗传学水平的异常变化使得DNA甲基化修饰具有重要的研究价值[20]。当然关于甲基化修饰在EM发病中的作用研究,还面临以下几个问题: (1) 通过反转异常甲基化修饰的基因表达来降低EM的发病率;(2) 在多个异常甲基化修饰的基因里面,查找某些与EM相关的特定基因;(3) 在EM发病总导致的基因异常甲基化修饰变化原因中,除了长期持续性的炎性刺激外还有其他原因导致基因的甲基化修饰异常还待研究。最后,本研究将为DNA甲基化修饰提供新的分子靶点,具有重要的EM的临床诊断和治疗参考价值。
【参考文献】
[1] Farquhar CM. Extracts from the “clinical evidence” Endometriosis[J]. BMJ, 2000,320: 1449-1452.
[2] Signorile PG, Baldi A. Endometriosis: new concepts in the pathogenesis[J]. INT J BIOCHEM CELL B,2010,42: 778-780.
[3] Rogers PA, D’Hooghe TM, Fazleabas A, et al. Defining future directions for endometriosis research: workshop report from the 2011 World Congress of Endometriosis In Montpellier, France[J]. Reprod sci, 2013,20: 483-499.
[4] Issa JP.Epigenetic variation and human disease[J]. J Nutr, 2002,132: 2388-2392.
[5] Nasu K, Kawano Y, Tsukamoto Y, et al. Aberrant DNA methylation status of endometriosis: epigenetics as the pathogenesis, biomarker and therapeutic target[J]. j obstet Gynaecol Res, 2011,37: 683-695.
[6] Slieker RC, Bos SD, Goeman JJ, et al.Identification and systematic annotation of tissue-specific differenti-ally methylated regions using the Illumina 450k array[J]. Epigenetics & chromatin, 2013,6: 26.
[7] Hoffmann MJ, Schulz WA. Causes and consequences of DNA hypomethylation in human cancer[J]. Biochemistry and cell biology Biochimie et biologie cellulaire, 2005,83: 296-321.
[8] Martini M, Ciccarone M, Garganese G, et al. Possible involvement of hMLH1, p16(INK4a) and PTEN in the malignant transformation of endometriosis. International journal of cancer[J]. IntJ Cancer, 2002,102: 398-406.
[9] Lokk K, Modhukur V, Rajashekar B, et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns[J]. Genome biol, 2014,15: r54.
[10] Wu Y, Halverson G, Basir Z, et al. Aberrant methylation at HOXA10 may be responsible for its aberrant expression in the endometrium of patients with endometriosis[J]. Am J Obstet Gynecol, 2005,193: 371-380.
[11] Hastings JM, Fazleabas AT. A baboon model for endometriosis: implications for fertility[J]. Reprod biol endo crin, 2006,4(Suppl 1): S7.
[12] Wu Y, Strawn E, Basir Z, et al. Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in women with endometri-osis[J]. Fertil Steril, 2007,87: 24-32.
[13] Parazzini F, Bertulessi C, Pasini A, et al. Determinants of short term recurrence rate of endome-triosis[J]. Eur J Obstet Gyn RB, 2005,121: 216-219.
[14] Derouich S, Attia L, Slimani O, et al.Medical treatment of endometriosis[J]. La Tunisie Med, 2015,93: 407-412.
[15] Kim JJ, Taylor HS, Lu Z, et al. Altered expression of HOXA10 in endometriosis: potential role in decidua-lization[J]. Mol Hum Reprod, 2007,13: 323-332.
[16] Lee B, Du H,Taylor HS. Experimental murine endometriosis induces DNA methylation and altered gene expression in eutopic endometrium[J]. Biol reprod, 2009,80: 79-85.
[17] Eun KH, Taylor HS. The role of HOX genes in human implantation[J]. Ann N. Y Acad of Sci, 2004,1034: 1-18.
[18] Lu H, Yang X, Zhang Y, et al. Epigenetic disorder may cause downregulation of HOXA10 in the eutopic endometrium of fertile women with endometriosis[J]. Reprod sci, 2013,20: 78-84.
[19] Naqvi H, Ilagan Y, Krikun G, et al. Altered genome-wide methylation in endometriosis[J]. Reprod sci, 2014,21: 1237-1243.
欢迎订阅2017年《同济大学学报(医学版)》杂志
《同济大学学报(医学版)》是由国家教委主管,同济大学主办,国内外公开发行的学术期刊。于1980年创刊(双月刊),刊号ISSN 1008-0392 /CN31-1901/R。
《同济大学学报(医学版)》被收录为“中国科技论文统计源期刊”(中国科技核心期刊);同时被解放军医学图书馆《中国生物医学期刊文献数据库CMCC》及《中国生物医学期刊引文数据库CMCI》收录。现有主要栏目: 专家笔谈、基础研究、临床研究、调查研究。综述栏目(有省部级以上的课题稿),每期128页。
全国各地邮局均可订阅《同济大学学报(医学版)》。邮发代号: 4-722。每期10元,全年60元。如当地订阅不便,编辑部可代办邮购。
来函请寄:
地址: 上海市杨浦区四平路1239号 同济大学学报(医学版)编辑部
邮编: 200092
电话: 021-65980705
E-mail: yxxb@tongji.edu.cn
DNA methylome analysis with methylation microarray in patients with Endometriosis
【Abstract】Objective To analyze DNA methylome in endometriosis patients with methylation microarray. Methods Tissue specimens of eutopic endometrium and ectopic endometrium were taken from 6 patients with endometriosis(EM) and normal endometrial specimens taken from 6 non-EM women. The genome methylation was detected with metylation microarray(Illumina Human Methylation450K Beadchip) The methylome profiling data were analyzed by clustering and principal component analysis(PCA). GO and KEGG pathway analysis was performed on the differentially expressed genes for biological functions. Results Our results showed that various epigenetic aberrations existed in endometriosis. The aberrant methylation genes showed different signature in eutopic endometrium and ectopic endometrium of EM patients, and normal endometrim by PCA analysis. Furthermore, GO and KEGG analysis showed that these genes were enriched in antigen processing and presentation and interferon-gamma-mediated signaling pathway, and involvement in Type 1 diabetes mellitus, allograft rejection, graft-versus-host disease, and autoimmune thyroid disease. Conclusion Endometrial DNA methylation abnormity has been identified as one specific feature of endometriosis as compared with normal. DNA methylation genes may provide useful biomarker targets for diagnostic and prognostic purposes.
【Key words】endometriosis; methylome; microarray; GO analysis; KEGG
收稿日期:2016-08-27
基金项目:上海浦东新区青年科技项目(PW2013B-4)
【中图分类号】R 713.7
【文献标志码】A
【文章编号】1008-0392(2016)06-0041-06