·基础研究·
【摘要】目的 探讨尼达尼布(Nintedanib)对抗血管紧张素Ⅱ(angiotensin Ⅱ, Ang Ⅱ)诱导的肾脏成纤维细胞产生细胞外基质的作用及其机制。方法 Ang Ⅱ处理大鼠肾脏成纤维细胞(NRK- 49F),Western 印迹法检测相关分子的蛋白表达量,DHE荧光探针法检测活性氧簇(ROS)的水平。结果 肾脏成纤维细胞中Ang Ⅱ通过诱导PI3K/Akt 信号通路的激活,引起纤维连接蛋白和胶原蛋白Ⅰ的积聚。Nintedanib能抑制Ang Ⅱ诱导的PI3K/Akt信号通路的激活。Nintedanib还可以抑制Ang Ⅱ引起的ROS水平升高。结论 Nintedanib可以抑制Ang Ⅱ诱导的ECM成分升高,其作用机制与其抑制PI3K/Akt激活并抑制ROS产生有关。
【关键词】尼达尼布; PI3K/Akt; 活性氧簇; 血管紧张素Ⅱ; 肾脏纤维化; 大鼠
肾脏纤维化是各种慢性肾脏疾病持续进展为终末期肾病的共同途径,主要标志为细胞外基质(extracellular matrixc, ECM)过度沉积,导致正常肾脏组织结构破坏及肾脏功能进行性下降。肾素-血管紧张素系统(renin-angiotensin system, RAS)的过度激活在肾脏纤维化的病理进程中有重要作用。作为RAS的最重要活性肽,血管紧张素Ⅱ(angiotensin Ⅱ, Ang Ⅱ)可引起与肾脏纤维化密切相关的细胞因子如TGF-β、TNF-α等的表达增多,并引起多条相关信号通路如TGF-β-Smads,MAPK,PI3K/Akt的激活[1]。
尼达尼布(Nintedanib)是一种三重酪氨酸激酶抑制剂,对血小板源生长因子(platelet derived growth factor, PDGF)、血管内皮生长因子(vascular endothelial growth factor, VEGF)和碱性纤维母细胞生长因子(basic fibroblast growth factor, bFGF)均有抑制作用[2]。初起主要用于肿瘤治疗[3-4],如肾癌、卵巢癌等,近年来发现Nintedanib对特发性肺间质纤维化(idiopathic pulmonary fibrosis, IPF)[5]有明显的疗效。本研究旨在探索Nintedanib对Ang Ⅱ引起的大鼠肾成纤维细胞细胞外基质过度增生的抑制作用,并探讨其作用机制。
1.1 试剂
DMEM高糖培养基购自美国Gibco公司;Nintedanib购自美国Selleck公司;Ang Ⅱ、坎地沙坦(candesartan, can)、LY294002、DPI、纤维连接蛋白(fibronectin, FN)一抗、超氧化物阴离子荧光探针二氢乙锭(dihydroethidium, DHE)购自美国Sigma公司;β-Actin一抗购自中国上海碧云天生物技术有限公司;Akt一抗、p-Akt一抗购自美国Cell Signaling公司;胶原蛋白 Ⅰ一抗购自美国Millipore公司。
1.2 细胞培养
NRK-49F细胞株用含10%FBS的DMEM高糖培养基,在37℃、5%CO2条件下培养、传代。用0.5% FBS的DMEM高糖培养基同步化24h后进行药物处理。给予细胞Ang Ⅱ(1μmol/L)刺激0、24、48h或者0、5、15、30、60、120min;加药处理细胞主要分为3组: 对照组(con);Ang Ⅱ组(1μmol/L);Ang Ⅱ(1μmol/L)+预处理坎地沙坦(10μmol/L)/Nintedanib(100nmol/L)/LY294002(10μmol/L)/DPI(10μmol/L) 1h组。
1.3 Western印迹法检测
以SDS细胞裂解液(加入蛋白酶抑制剂)提取细胞总蛋白,BCA法测定蛋白浓度。40μg蛋白上样,行SDS-PAGE电泳,转膜至PVDF膜。TBST冲洗,脱脂奶粉封闭2h。然后将膜放入稀释的一抗(按照说明书),4℃孵育过夜。次日,TBST冲洗后,将膜放入相应的辣根过氧化物酶标记的二抗(稀释倍数均为1∶1000),室温孵育2h,TBST冲洗后,用凝胶成像系统拍照,Smart viewer软件进行灰度分析,内参照为β-actin。
1.4 ROS水平检测
利用DHE荧光探针检测方法反映细胞内超氧阴离子(·O2-)的水平,DHE脱氢后和RNA或DNA结合产生红色荧光。Ang Ⅱ处理NRK-49F细胞24h后,用10μmol/L DHE在37℃下孵育细胞30min进行荧光探针装载,用PBS洗涤3次后在荧光显微镜下观察细胞内荧光强度变化。
1.5 统计学处理
实验所得计量资料以![]() ±s表示,利用SPSS 17统计分析软件,多组间两两比较采用单因素方差分析(one-way ANOVA),两组间的比较采用t检验,P<0.05为差异有统计学意义。
±s表示,利用SPSS 17统计分析软件,多组间两两比较采用单因素方差分析(one-way ANOVA),两组间的比较采用t检验,P<0.05为差异有统计学意义。
2.1 Nintedanib抑制Ang Ⅱ诱导的细胞外基质合成
NRK-49F细胞株中,给予Ang Ⅱ(1μmol/L)刺激0、24、48h后,Ang Ⅱ引起胶原蛋白 Ⅰ、纤维连接蛋白水平明显升高,其作用强度呈时间依赖性,见图1。
用AT1R阻断剂坎地沙坦(10μmol/L)预处理细胞1h后,再给予Ang Ⅱ刺激48h,Western 印迹法结果显示坎地沙坦显著抑制Ang Ⅱ诱导的胶原蛋白 Ⅰ、纤维连接蛋白水平升高,见图2。
用Nintedanib(100nmol/L)预处理细胞1h后,再加入Ang Ⅱ(1μmol/L) 刺激细胞48h,Western 印迹法检测结果显示,Nintedanib明显抑制Ang Ⅱ 引起胶原蛋白 Ⅰ、纤维连接蛋白水平升高,见图3。
2.2 Nintedanib抑制Ang Ⅱ所诱导的Akt磷酸化
Ang Ⅱ(1μmol/L)刺激48h NRK-49F细胞后,在0、5、15、30、60、120min,Western 印迹法检测Akt磷酸化水平,结果显示,细胞Akt磷酸化明显增强,见图4。
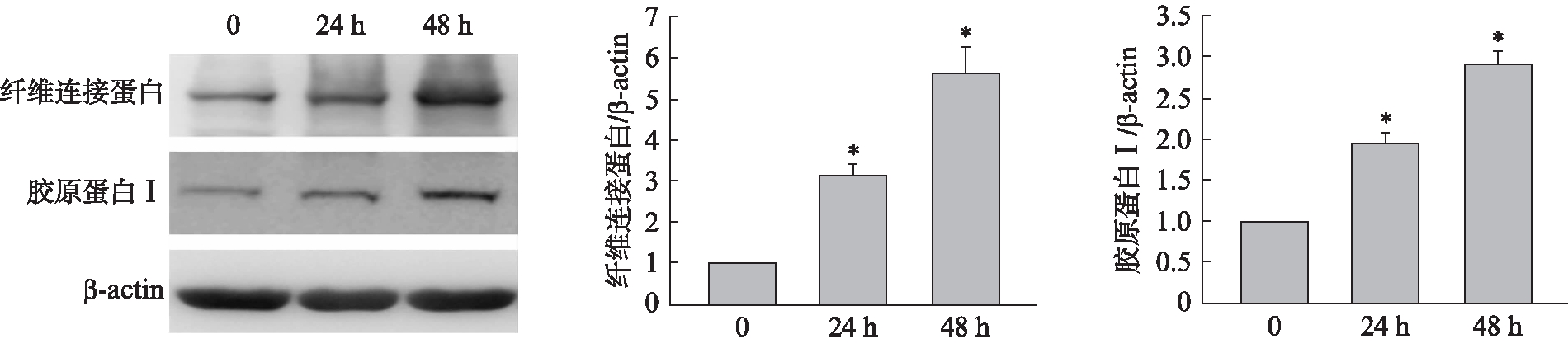
图1 Ang Ⅱ诱导细胞外基质合成增加
Fig.1 Ang Ⅱ induced the synthesis of extracellular matrix
与Ang Ⅱ处理细胞0h相比,*P<0.01,n=3
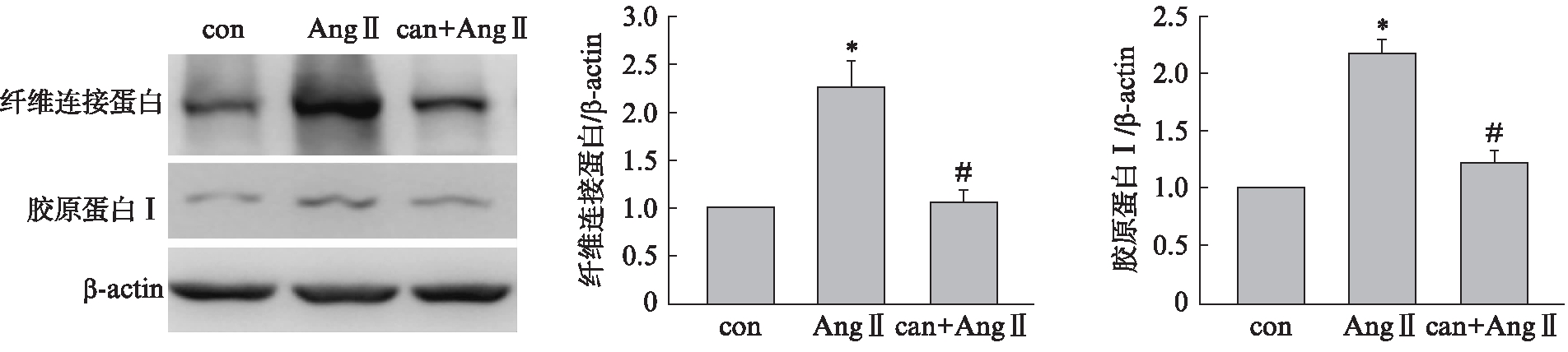
图2 坎地沙坦阻断Ang Ⅱ诱导的细胞外基质合成增加
Fig.2 Candesartan blocked the synthesis of extracellular matrix induced by Ang Ⅱ
与con组相比,*P<0.01,与Ang Ⅱ组相比,#P<0.01;n=3
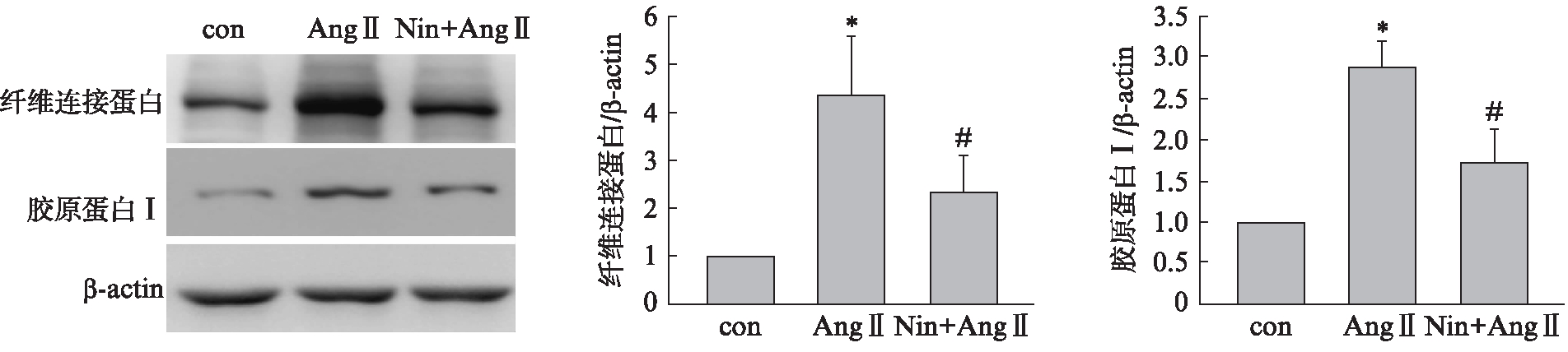
图3 Nintedanib阻断Ang Ⅱ诱导的细胞外基质合成增加
Fig.3 Nintedanib blocked the synthesis of extracellular matrix induced by Ang Ⅱ
与con组相比,*P<0.01,与Ang Ⅱ组相比,#P<0.01;n=3
给予PI3K/Akt抑制剂LY294002(10μmol/L)预处理细胞1h,再给予Ang Ⅱ(1μmol/L)刺激48h,Western印迹法结果显示LY294002可以抑制Ang Ⅱ诱导的胶原蛋白 Ⅰ、 纤维连接蛋白水平明显升高,见图5。
Nintedanib(100nmol/L)预处理细胞1h后,给予Ang Ⅱ(1μmol/L)刺激0、5、15、30、60、120min,收集细胞蛋白,Western印迹法结果显示,Nintedanib 明显抑制Ang Ⅱ引起的Akt磷酸化水平升高,见图6。
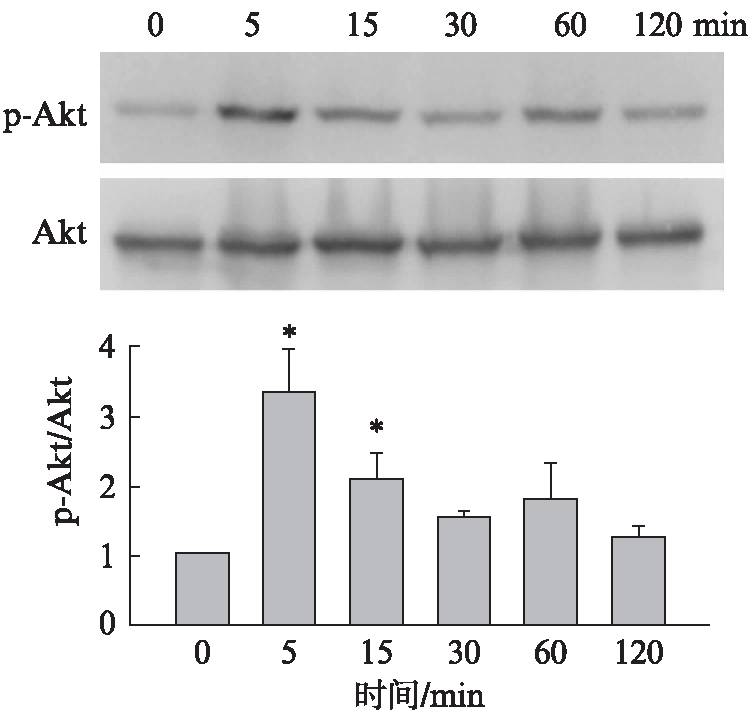
图4 Ang Ⅱ激活Akt信号通路
Fig.4 Ang Ⅱ induced the activation of Akt signaling
与Ang Ⅱ作用0min组相比,*P<0.01;n=3
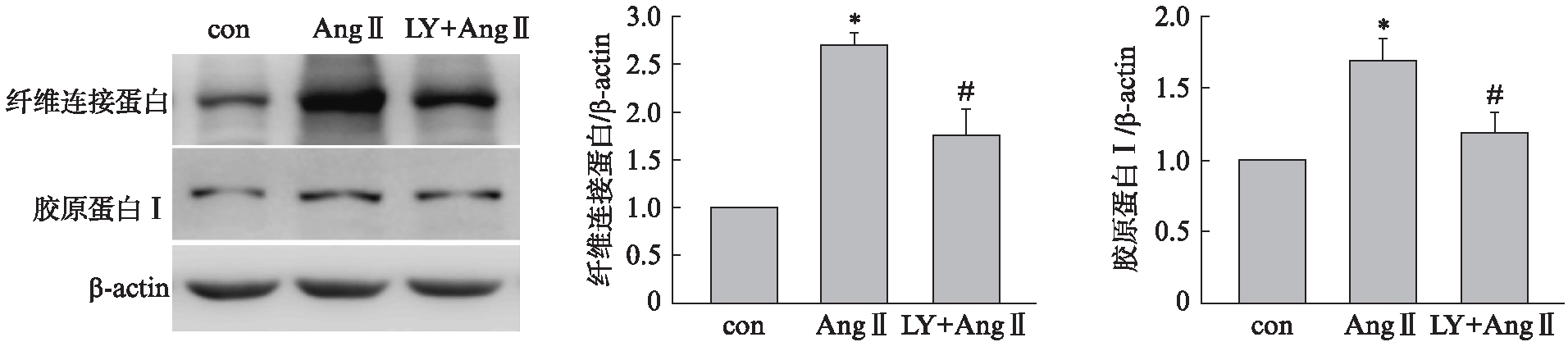
图5 LY294002阻断Ang Ⅱ诱导的细胞外基质合成增加
Fig.5 LY294002 blocked the synthesis of extracellular matrix induced by Ang Ⅱ
与con组相比,*P<0.01,与Ang Ⅱ组相比,#P<0.01;n=3
2.3 Nintedanib抑制Akt信号通路激活引起ROS水平增高
Ang Ⅱ(1μmol/L)处理细胞24h后,利用DHE荧光探针检测细胞内ROS的水平。荧光结果显示,与对照组相比,Ang Ⅱ处理组的ROS水平明显升高,见图7。

图6 Nintedanib阻断Ang Ⅱ诱导的Akt激活
Fig.6 Nintedanib blocked the activation of Akt induced by Ang Ⅱ
与con组相比,*P<0.01,与Ang Ⅱ组相比,#P<0.05,##P<0.01;n=3
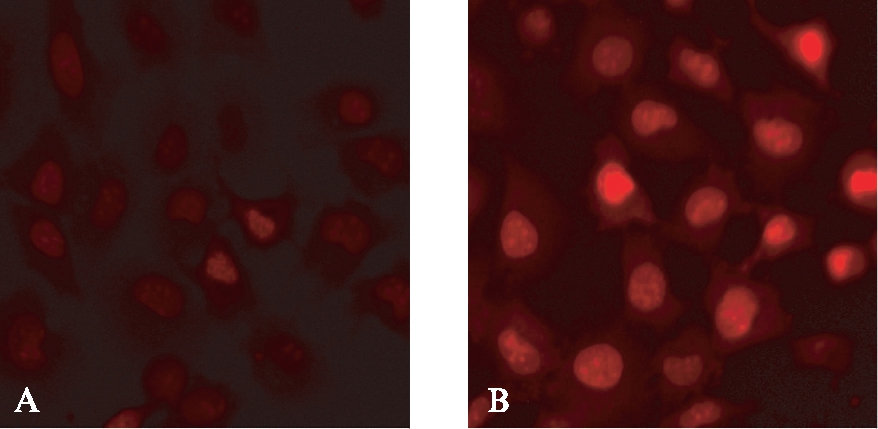
图7 Ang Ⅱ诱导ROS水平增高
Fig.7 Ang Ⅱ induced the production of ROS(DHE,×400)
A: con组; B: Ang Ⅱ组
给予PI3K/Akt抑制剂LY294002(10μmol/L)预处理细胞1h,再给予Ang Ⅱ(1μmol/L)刺激24h,结果显示LY294002可以抑制Ang Ⅱ诱导的ROS水平升高。Nintedanib(100nmol/L)预处理细胞1h后,给予Ang Ⅱ刺激24h,利用DHE荧光探针检测细胞内ROS的水平。结果表明,Nintedanib可显著抑制Ang Ⅱ所引起的ROS水平升高,见图8。
给予NADPH氧化酶抑制剂DPI(10μmol/L)预处理细胞1h,再给予Ang Ⅱ(1μmol/L)刺激48h,Western印迹法显示DPI可以显著抑制Ang Ⅱ诱导的胶原蛋白 Ⅰ、纤维连接蛋白水平升高,见图9。
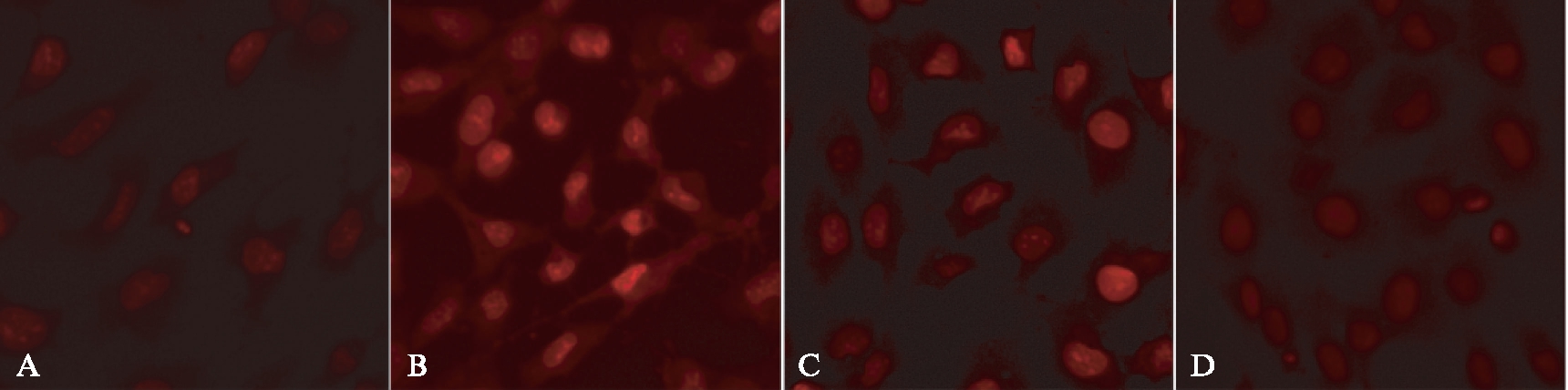
图8 LY294002及Nintedanib均可抑制Ang Ⅱ引起的ROS水平升高
Fig.8 LY294002 and Nintedanib inhibited ROS production induced by Ang Ⅱ (DHE,×400)
A: 对照组;B: Ang Ⅱ组;C: LY+Ang Ⅱ组;D: Nin+Ang Ⅱ组

图9 DPI阻断Ang Ⅱ诱导的细胞外基质合成增加
Fig.9 DPI blocked the synthesis of extracellular matrix induced by Ang Ⅱ
与con组相比,*P<0.01,与Ang Ⅱ组相比,#P<0.01;n=3
目前,已有的关于Nintedanib抗纤维化机制研究均是针对其对肺脏纤维化的拮抗机制。体外实验结果表明,Nintedanib可抑制PDGFR-α,PDGFR-β的激活和正常人肺成纤维细胞的增殖,也可抑制IPF患者来源的肺成纤维细胞的增殖[6]。Nintedanib可抑制IPF患者肺成纤维细胞中PDGF或FGF-2引起的细胞迁移,同时可抑制TGF-β诱导的成纤维细胞-肌纤维母细胞转化。另外,Nintedanib可增加MMP-2的活性,抑制TIMP-2及TGF-β引起的ERK通路激活,从而抑制肺成纤维细胞中ECM成分的表达。体内实验表明,Nintedanib可抑制小鼠肺组织中血小板衍生生长因子受体(platelet-derived growth factor receptor, PDGFR)的激活及其下游信号通路。在两种不同的小鼠IPF模型中,Nintedanib可导致肺组织中胶原蛋白沉积明显减少并有抗炎作用[7]。
本研究显示,Nintedanib能抑制Ang Ⅱ刺激所引起PI3K/Akt通路的激活,使用Nintedanib或PI3K/Akt抑制剂LY294002均明显降低了Ang Ⅱ诱导的ECM合成增多。
PI3K/Akt为细胞内重要的信号通路,在多种细胞进程如增殖、分化和凋亡中发挥重要作用[8]。PI3K/Akt信号通路与包括肾脏在内的多种器官纤维化的形成密切相关。在肾小球系膜细胞中,给予PI3K/Akt抑制剂LY294002,可明显抑制TGF-β引起的Ⅰ型胶原表达[9]。本研究中,Ang Ⅱ刺激可引起PI3K/Akt通路的激活并导致细胞外基质增多,给予PI3K/Akt抑制剂LY294002后明显降低了Ang Ⅱ诱导的ECM增多,再次证实了PI3K/Akt通路参与肾脏成纤维细胞ECM增多。
PI3K/Akt信号通路的激活需要一系列受体酪氨酸激酶的参与,如IGF-1R、HER2/Neu、VEGFR、PDGFR等[10]。活性氧簇(ROS)在维持细胞的正常生理功能中起重要作用,ROS可激活多条与肾脏纤维化相关的信号通路。NADPH氧化酶被认为是ROS的主要来源之一,研究证实Ang Ⅱ是激活NADPH氧化酶的主要刺激因子。这也是Ang Ⅱ导致肾脏纤维化的重要机制之一[11]。在病变肾脏中ROS的产生增多很大程度上是由于肾脏中RAS系统的过度激活[12]。Ang Ⅱ通过与其Ⅰ型受体(AT1R)结合,激活NADPH氧化酶,产生ROS,从而参与机体功能的调节。目前的研究发现Ang Ⅱ激活NADPH氧化酶主要有以下三条途径: 一是PKC途径,二是c-Src激酶途径;三是受体酪氨酸蛋白激酶途径: 即当Ang Ⅱ与AT1R结合后,激活PDGFR、EGFR、IGFR,进而激活酪氨酸蛋白激酶,并与PI3K的SH2结构结合,使Rac活化并移位到细胞膜,从而促进NADPH氧化还原反应平台的组装,产生大量ROS。
在本研究中,Ang Ⅱ 可诱导肾脏成纤维细胞的ROS水平明显升高,给予NADPH氧化酶抑制剂DPI则可显著抑制ECM蛋白的产生,同样给予Nintedanib也可以明显抑制Ang Ⅱ 诱导的ROS产生并减少ECM产生。由此认为,Nintedanib作为酪氨酸激酶抑制剂,抑制了Ang Ⅱ 所诱导的酪氨酸蛋白激酶的激活,进而抑制后者与PI3K的Sh2结构结合,阻止Rac移位到胞膜,使NADPH氧化酶受到抑制,ROS产生明显减少,最后减轻肾脏成纤维细胞ECM沉积。
本研究观察了Nintedanib对肾脏细胞过度表达ECM的拮抗作用,为开展Nintedanib作为治疗肾脏纤维化药物的研究提供了理论依据,但还需要在后续研究中进行体内研究并对其作用机制做更深入的研究。
【参考文献】
[1] Liu GX, Li YQ, Huang XR, et al. Disruption of Smad7 promotes ANG Ⅱ-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF.kappaB-dependent mechanisms in mice[J]. PLoS One, 2013,8(1): e53573.
[2] Hilberg F, Roth G J, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy[J]. Cancer Res, 2008,68(12): 4774-4782.
[3] Eisen T, Shparyk Y, Macleod N, et al. Effect of small angiokinase inhibitor nintedanib(BIBF 1120) on QT interval in patients with previously untreated, advanced renal cell cancer in an open-label, phase Ⅱ study[J]. Invest New Drugs, 2013,31(5): 1283-1293.
[4] Vergote I. Novel therapies, including enzastaurin, in the treatment of ovarian cancer[J]. Expert Opin Investig Drugs, 2014,23(5): 579-598.
[5] Wollin L, Maillet I, Quesniaux V, et al. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis[J]. J Pharmacol Exp Ther, 2014,349(2): 209-220.
[6] Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis[J]. Respir Res, 2014,15(1): 157.
[7] Wollin L, Maillet I, Quesniaux V, et al. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis[J]. J Pharmacol Exp Ther, 2014,349(2): 209-220.
[8] Wang J, Chu ES, Chen HY, et al. microRNA-29b prevents liver fibrosis by attenuating hepatic stellate cell activation and inducing apoptosis through targeting PI3K/AKT pathway[J]. Oncotarget, 2015,6(9): 7325-7338.
[9] Runyan CE, Schnaper HW, Poncelet AC. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-beta1[J]. J Biol Chem, 2004,279(4): 2632-2639.
[10] Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development[J]. Curr Cancer Drug Targets, 2004,4(3): 235-256.
[11] Nakanishi A, Wada Y, Kitagishi Y, et al. Link between PI3K/AKT/PTEN Pathway and NOX proteinin diseases[J]. Aging Dis, 2014,5(3): 203-211.
[12] Kashihara N, Haruna Y, Kondeti VK, et al. Oxidative stress in diabetic nephropathy[J]. Curr Med Chem, 2010,17(34): 4256-4269.
Nintedanib suppresses overexpression of extracellular matrix induced by angiotensin Ⅱ in renal fibroblasts
【Abstract】Objective To investigate the effect of nintedanib on extracellular matrix production induced by angiotensin Ⅱ(Ang Ⅱ) in renal fibroblasts and related mechanism. Methods Renal fibroblast cells were treated with Ang Ⅱ. Expression of fibronectin, collagen Ⅰ levels was determined by Western blotting, and the ROS level was detected by DHE fluorescent probe. Results Ang Ⅱ induced the phosphorylation of PI3K/Akt signalling, with the accumulation of fibronectin and collagen Ⅰ in renal fibroblast cells. Nintedanib blocked the phosphorylation of Akt and attenuated the increase of fibronectin and collagen Ⅰ induced by Ang Ⅱ. Nintedanib also inhibited Ang Ⅱ induced ROS generation. Conclusion Nintedanib can attenuate the accumulation of ECM proteins induced by Ang Ⅱ, which may be associated with the suppression effect on PI3K/Akt signalling activation and subsequent ROS overproduction.
【Key words】Nintedanib; PI3K/Akt; reactive oxygen species; angiotensin Ⅱ; renal fibrosis; rat
doi:10.16118/j.1008-0392.2015.04.003
收稿日期:2015-02-26
基金项目:国家自然科学基金(81370790)
【中图分类号】R 34
【文献标志码】A
【文章编号】1008-0392(2015)04-0013-06